
We use cookies to ensure our website works properly and to personalise your experience. Cookies policy
Shri K R Pandav Institute of Pharmacy, Nagpur
Sedentary lifestyle has emerged as a major public health concern worldwide and is strongly associated with cardiovascular diseases, particularly atherosclerosis. Rapid urbanization, technological advancement, and changes in occupational patterns have led to a significant reduction in daily physical activity. Atherosclerosis is a chronic inflammatory disease of the arterial wall and represents the underlying pathology of most cardiovascular disorders. The present review aims to comprehensively examine the relationship between sedentary lifestyle and the development and progression of atherosclerosis, with emphasis on underlying pathophysiological mechanisms and preventive strategies. A narrative review of the literature was conducted using electronic databases such as PubMed, Google Scholar, and Scopus. Articles published between 2000 and 2024 were reviewed using keywords including sedentary lifestyle, physical inactivity, atherosclerosis, endothelial dysfunction, lipid profile, nitric oxide, and oxidative stress. Evidence indicates that prolonged physical inactivity promotes endothelial dysfunction, dyslipidemia, oxidative stress, and chronic inflammation, whereas regular physical activity improves vascular function and lipid metabolism. In conclusion, sedentary lifestyle is an independent and modifiable risk factor for atherosclerosis. Lifestyle modification through regular physical activity, along with appropriate pharmacological interventions, plays a crucial role in preventing and managing atherosclerotic cardiovascular disease.
Sedentary lifestyle is an evident factor which affects cardiovascular health. About 40% of adults allegedly get less than 10 minutes of persistent physical activity every week. This prolonged lack of physical activity leads to metabolic issues and facilitates the processes that triggers a disease conditions like atherosclerosis. Evidence indicates that insufficient physical activity is an independent risk factor for cardiovascular disease, promoting endothelial dysfunction, lipid accumulation, and systemic inflammation—key mechanisms in the initiation and progression of atherosclerotic plaque. Thus, sedentary behavior represents a direct and modifiable contributor to the global burden of atherosclerosis, emphasizing the importance of physical activity promotion in preventive cardiology.
Sedentary behaviour is extensively seen as a significant lifestyle factor which leads to metabolic and cardiovascular diseases. Which includes a slight physical activity or any kind of exercise, commonly consisting of hours of sitting or enervated tasks like reading, desktop jobs or using electronic devices such as mobile phone, televisions or computers. Today’s lifestyle has markedly reduced daily physical activity levels. Latest-occurring study shows approximately 50% drop in physical activity in countries like the Unites Kingdom and United States. This reduction has been closely associated with the rising prevalence of diabetes and cardiovascular disease (CVD).
Sedentary lifestyles are physiologically destructive as reduction in muscular activity leads to improper blood flow, also causes gravitational pooling of blood into the lower body veins, increases arterial pressure, reduction in vascular sheer stress arising from the friction of blood with vessel endothelial layers. All the above changes facilitate endothelial dysfunction, a core mechanism behind the progression of CVD. Meanwhile, the reduced shear stress lowers nitric oxide (NO) production leading to lower NRF2 antioxidant pathway activation, the NO as well as NRF2 both play a major role in vascular protection. All such vascular imbalances that occur due to the lack of physical activity (sedentary lifestyles) stimulates the production of reactive oxygen species (ROS) that oxidizes low-density lipoprotein cholesterol (LDL-C) along with this it promotes vascular inflammation and excessive oxidative stress production. All these factors are progressive agents of vascular remodeling and various cardiovascular disease (CVD) pathogenesis.
Atherosclerosis triggers major cardiovascular disorders such as coronary artery disease and peripheral artery disease. It is a disease condition that arises mainly from elevated plasma cholesterol level stimulating the formation of plaques within the arterial wall which are termed as lipid -rich atherosclerotic plaques. Such plaques sometimes become unstable and when disturbed releases their thrombogenetic elements into circulating blood, which triggers acute clinical events such as myocardial infarction, unstable angina, ischemic stroke, and sudden cardiac death. Development of atherosclerosis within the vascular system is not uniform as only few tender and susceptible plaques develop and others remain clinically non-symptomatic and silent throughout life. The individual remains asymptomatic until one such vulnerable plaque erupts or ruptures releasing thrombosis that obstruct blood flow ultimately life-threatening ischemic syndrome and strokes.
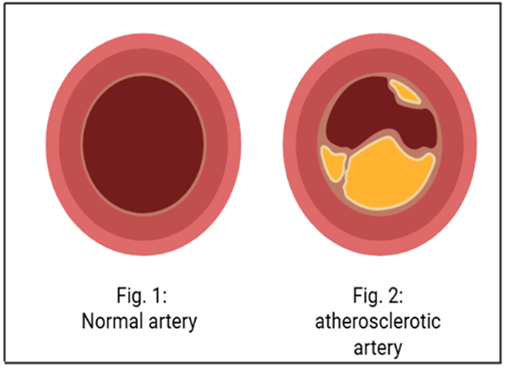
PATHOPHYSIOLOGY OF ATHEROSCLEROSIS
Atherosclerosis is function of hyperlipidemia and lipid oxidation. A major part of mortality rate in develop countries are due to atherosclerosis. It is obtained from Greek word “athereosis” and “sclerosis” which collectively means thickening of intimal layer of arteries and accumulation of fat.
“Athereosis” – Accumulation of fat accompanied by several macrophage and “sclerosis” – Fibrosis layer of smooth muscle cell and leucocytes.
It starts when endothelium becomes damaged allowing the harmful type of cholesterol to build up on the artery wall body sends white blood cells to clean up the cholesterol, but sometimes cells get stuck at affected site and plaque builds up made up of cholesterol, macrophages, Calcium and other substances from the blood.
Atherosclerotic plaque is of two types:
Sometimes plaque eventually breaks open. In this condition, platelets gather in the affected area and can stick together, forming blood clots. This can block the artery, leading to life threating complications.
Cellular Components of Atherosclerosis
Atherosclerosis is a prolonged immunoinflammatory, and fibroproliferative disorder of the arterial wall that firstly influences medium and large-sized arteries. It is a Polygenic disorder characterized by lipid build-up, inflammatory cell infiltration, endothelial dysfunction, significantly leading to formation of atherosclerotic plaque.
The crucial cellular players in the pathophysiology of atherosclerosis include:
Among these, endothelial cells are of specific significance because they form the first obstacle between the circulating blood and the vascular wall. Their disruption is commencing step in the sequence of events leading to atherosclerosis.
Endothelial dysfunction initiates the development of a vascular state susceptible to lipid infiltration, inflammation and plaque formation. The areas in artery which are severely exposed to either disturbed blood flow or hemodynamic stress become lesion prone. Even the leaky, activated and dysfunctional endothelial cell layer appears to be intact.
This endothelial dysfunction promotes easy vascular permeability of lipoprotein (LDL) and plasma proteins into the sub-endothelial space, loss of anti- thrombotic and anti- inflammatory properties and reduction in nitric oxide (NO) production disturbing vasodilation.
Progression of the damage causes localized removal of endothelial cells and exposing sub-endothelial layer to shocks. This matrix then becomes site for platelet adhesion and aggregation initiating development of atherosclerotic lesions.
Low-density Lipoprotein (LDL) enters the lesion prone subendothelial space and gets trapped into the extracellular proteoglycans. Trapped LDL undergoes oxidation by reactive oxygen species (ROS) and lipoxygenase or myeloperoxidase forming oxidizes LDL (Ox-LDL) which is cytotoxic to endothelial cells attracting monocytes from blood to the lesion site and promotes adhesion of leukocytes[2].
Nitric oxide (NO) plays a major role in vasodilation, platelet aggregation inhibition and leucocyte adhesion. Nitric oxide acts as both protective and damaging agent to the endothelial cells depending upon its source of production. NO produced by endothelial cells are anti- atherogenic and protective. On contrary, NO released from macrophages in inflammation conditions are oxidative which causes oxidative damage to LDL making it pro-atherogenic.
Collectively, endothelial dysfunction hinders vascular relaxation, maintaining pro-thrombotic and pro-inflammatory conditions which ultimately leads individual to clinical events like myocardial infraction or stroke developed by plaque progression and plaque rupture.
Thus, management strategies of Atherosclerosis mainly aim to promote nitric oxide (NO) bioavailability and impart LDL oxidation.
Leukocytes specially monocytes and macrophages plays a crucial role in the induction, progression and consequences of atherosclerosis. Their interrelations with lipids, endothelial cells and cytokines form a self-maintaining inflammatory cycle, leading to growth of plaque and eventual breakdown. This non-functional endothelium produces adhesion molecules (such as VCAM-1 and ICAM-1) and secretes chemotactic cytokines and recruit leukocytes to the arterial wall.
Two important chemotactic agents are:
These two molecules recruit T cells and monocytes from bloodstream to the inflammatory locus.
Once monocytes attach to the endothelium, they emigrate into the intima (inner layer of the vessel wall) where they separate into macrophage under the impact of cytokines such as M-CSF (macrophage colony-stimulating factor). Macrophages are significant immune cells which swallow and digest cell debris, lipids and pathogens through a process called phagocytosis. Inside the intima, macrophages swallow Ox-LDL particles through scavenger receptors such as SR-A and CD36. In contrast to the normal LDL receptor, these scavenger receptors are not suppressed by cellular cholesterol levels, so macrophages persist to take up the lipids substantially. As a result, macrophages aggregate cholesterol esters, transforming into foam cells.
Smooth muscle cells (SMCs) from the arterial wall begin to involve actively. They are immersed in a fibroproliferative response, which means they proliferate and generate fibrous tissue as part of body’s healing and repair mechanism after arterial injury.
When the intima is impaired, SMCs migrate from the middle layer (media) to inner layer and start recovering the injury by:
This operation helps regulate the artery and prevent proliferation at first. If damaging factors like smoking, high cholesterol or high blood pressure persist for many years.
This coagulates arterial wall and constrict the lumen.
PROCESS OF ATHEROSCLEROSIS
This process includes three steps which are:
Atherosclerosis starts with the formation of fatty streaks, the primary visible lesions caused by the aggregation of cholesterol-rich LDL in the inner lining of arteries. LDL permeates the endothelium and attaches to proteoglycans, specifically heparan sulphate, which facilitates its accumulation and shows excision, leading to an escalating buildup. Monocytes immigrate into the endothelial layer, convert into macrophages, and phagocytose LDL to form foam cells, while SMCs replicate and extracellular fat deposits aggregate, generating a soft lesion. Over a period of time, the plaque dilated, at first amplifying outward but ultimately constricting the arterial lumen; once it includes more than 40% of the inner elastic layer, the vessel is regarded substantially obstructed[3]. This development sets the stage for advanced atherosclerotic disease and potential health complications such as myocardial infarction, angina or stroke.
Low-density lipoprotein-cholesterol [LDL-C] Trapping
LDL cholesterol [LDL-C] becomes trapped, marking the first step of atherogenesis. Normally blocked by tight endothelial junction, LDL can still enter endothelial cells via endocytosis. In normal condition, a dynamic equilibrium exists between plasma LDL level and LDL content within the arterial wall. When blood lipid level increase more LDL become trapped in the intima, primarily through interaction with proteoglycans that strongly attract LDL. This buildup disturbs the equilibrium, resulting in lipid retention within the arterial wall. Since LDL trapping increases with blood LDL, elevated serum LDL indicates atherogenic risk.
Activation of endothelial cell
Altered LDL promotes endothelial cell to produce adhesion molecule [such as ICAM-1, E- selection, VCAM-1][3]. Circulating immune cell including monocyte and T-lymphocytes are adhere on the endothelial layer through these adhesion molecule. Chemokines secrete by endothelial cells including MCP-1, which attached monocytes from circulation into arterial intima. Therefore, the endothelial transition from defensive function to an inflammatory and adhesive state.
Leukocyte activation
Macrophages are mature, when monocytes are migrated under the endothelium. Macrophages are bind & Adopt oxidized LDL by which scavenger receptors (it's does not regulated like LDL receptors, which is why uptake continues persistent). perpetuating endothelial dysfunction and engaged additional immune cells, when initialize macrophages produce inflammatory cytokines and within the arterial wall it's leads to a self-perpetuating inflammatory cycle.
Foam cell formation
The accumulation of foam cells in the arterial walls results in the formation of fatty streaks, an early sign in the development of atherosclerosis.
The macrophages produced in intima help in removal of extracellular lipid by accumulating LDL. Lipid accumulated macrophage then leaves the arterial wall through the lipids out of arterial wall area. For a normal Artery rate of lipid entry and removal are proportional, dysfunction of this mechanism leads lipid accumulation in arterial intima, accelerating rate of atheroma formation. The scavenger receptors present on macrophages act as ox- LDL binding site, enhanced by cytokinin, oxidizes low-density Lipoprotein and macrophages colony-stimulating factors.
As the macrophages engulf ox- LDL they become foam cells, yellowish lipid loaded macrophages. Over the time foam cells in the developing intima die by apoptosis, developing lipid - rich necrotic core in the center of atherosclerotic plaque, increasing severity of the disease[3].
Damaged vascular tissue marks the beginning of atheroma formation, leading to the release of signalling molecules from smooth muscle cells and endothelial cells such as cytokinin, interleukin-1 (IL-1) and tumour necrosis factor (TNF). All such components promote migration of SMC towards inner side of the blood vessel wall where they produce extravascular matrix components (ECM). This accumulated matrix material and cells form a structure named as fibrous cap. Fibrous cap is composed of collagen- rich fibre tissues, SMC, macrophages and T lymphocytes. The above components together turn into a mature atherosclerotic plaque, which situates into vessel's lumen and obstructs blood flow.
Structural stability of the fibrous cap is compromised and it weakens due to the lysis of extracellular matrix by enzymes called as metalloproteinases released from macrophages. On other hand TNF-α released by T cells lead to inhibition of collagen synthesis in SMC. As a result, fibrous cap repair and healing delayed as well as decreased. All over ECM degradation and collagen reduction further give birth to plaque rupture process initiation. On rupturing, the fibrous cap components such as collagen and lipids get released into bloodstream, promoting platelet adhesion and formation of blood clot that blocks the blood flow producing life-threatening severe cardiovascular events like stroke or heart attack.
Atherosclerotic plaques build up sequentially within the artery walls as a result of inflammation and injury. The accumulation initiates with changes in the endothelium, which interacts with macromolecules and components of the blood. This interaction amplifies the transfer of proteins into the plasma, facilitating to the initial stages of plaque buildup. The arterial smooth muscle plays a significant role in regulating vascular repair and in metabolizing blood products such as lipids. It also releases various cytokines, which helps in controlling the tone and stability of vascular wall.
Various types of cells, including lymphocytes, take part in inflammatory and immune response, as the plaque persists to develop. The crux of the plaque, also called as lipid nucleus, consists of calcium deposits, cholesterol esters, foam cells, cellular debris and a mixture of fatty substances.
This core of lipid emerges as a pale-yellow mass, and this yellowish colour of lipid core is because of the carotenoid pigments. Overall, plaque has fibrous outer layer for stability and the lipid-rich nucleus or core at the centre, which simultaneously can lower blood flow, constrict the artery and increase the risk of rupture directing to heart attack or stroke.

EFFECT OF PHYSICAL INACTIVITY ON HDL, TRIGLYCERIDES AND LDL
Physical inactivity has a negative impact on HDL commonly called as good cholesterol in human body. HDL plays a prophylactic role by transporting elevated cholesterol from the bloodstream and vessel walls to the liver for elimination. With irregular exercise, the body generates less HDL, and the HDL particles that are present become less efficient at removing cholesterol. As HDL levels lowers, the ability of body to clear adverse LDL from arteries also weakens. This permits more cholesterol to aggregate inside blood vessels, increasing formation of plaque and increasing the risk of conditions like atherosclerosis, heart disease and stroke.
Physical inactivity has a negative impact on triglyceride levels in human body. When a person is physically inactive, gives rise to high levels of triglyceride, resulting triglycerides to aggregate in the bloodstream. The lowered activity of an enzyme called lipoprotein lipase (LPL), which helps in degradation of triglycerides, causes higher level of these fats in the blood. As a result, residual triglycerides are stored in fat cells, resulting in weight gain and abdominal fat aggregation. Physical inactivity also facilitates insulin resistance, causing the liver to generate more triglycerides and reducing body’s ability to metabolize fats. Furthermore, a sedentary lifestyle decreases the metabolic rate, which means sugars and fats are not effectively used for energy, which further increases triglyceride levels. Over time, excess triglycerides can lead to plaque formation in blood vessels, increasing the risk of atherosclerosis, heart disease and stroke.
Physical inactivity has a significant effect on the levels of low-density lipoprotein (LDL) in the human body. LDL, commonly called as “bad cholesterol”, contributes to transporting cholesterol to various tissues; however, when the concentration of LDL increases, it leads to the buildup of fatty deposits or plaques within arterial walls, a process known as Atherosclerosis. Lack of regular exercise or physical activity decrease the body’s ability to deploy and metabolize fats effectively.
Leading to an elevate in circulating LDL levels. Moreover, physical inactivity is correlated with the generation of smaller and more concentrated LDL particles, which are more atherogenic- which means they can more efficiently infiltrate the arterial lining and cause damage and inflammation. Physical inactivity also reduces the activity of enzymes such as lipoprotein lipase, which are crucial for the breakdown and clearance of LDL from the bloodstream. Over time, this causes cholesterol aggregation, contraction of arteries and increased cardiovascular risk.
PHYSICAL ACTIVITY AND PREVENTION OF ATHEROSCLEROSIS
How physical activity affects LDL
Both the quantity and quality of LDL are influenced by regular physical activity. Aerobic exercises such as brisk walking or cycling increase the activity of LDL receptors in liver and leads to reduced amount of LDL in circulation. Physical activity also decreases the production of VLDL (very low-density lipoprotein) which is a precursor of LDL. As a result, physical activity improves LDL to HDL levels.
Physical activity on other hand improves antioxidant production of body such as enzymes like superoxide dismutase and glutathione peroxidase become more active and reduces the LDL oxidation. Thus, physical activity having potential of lowering both the amount and atherogenic ability of LDL protect arteries from damage.
How physical activity Enhances Nitric oxide (NO)
During exercise, blood flow rises, which increases the force of blood pushing against the inner walls of the arteries known as shear stress. This physical signal turns on eNOS in endothelial cell through the PI3K/AKt pathway, resulting in more nitric oxide being produced. At the same time, exercise increases calcium level inside the cells, which help activates eNOS through the calcium-calmodulin complex. With consistent training, these effect become lasting: exercise enhance eNOS gene activity, raises the count of endothelial progenitor cells that repair blood vessels & increases the regular production of nitric oxide[4].
Exercise lowers oxidation stress, so there are fewer free radicals to break down the nitric oxide. This helps nitric oxide stay active longer, allowing it to protect blood vessels. As a result, the endothelium stays healthy, blood flows betters, & reduce risk of atherosclerotic damage.
Interrelation between LDL, NO and Endothelial disfunction
When LDL and specially oxidized LDL accumulate in the arteries, they impair the endothelium and intervene in eNOS activity. This decrease NO2 production and triggers endothelium disfunction. Without enough NO, blood vessels become firm and constricted, platelets begin to stick together, and there is increase in inflammation-all of these increases further plaque formation. Thus, high LDL and low NO form a detrimental cycle: LDL causes endothelial impairment, and endothelium-dysfunction allows more LDL to aggregate.
Physical activity helps break this vicious cycle. By decreasing LDL levels and oxidation while simultaneously rising NO production, exercise repairs endothelial balance and prevents the progression of atherosclerosis.
How Physical activity prevents Atherosclerosis progression
Exercise prevents blood vessels in multiple ways. It decreases LDL and oxidized LDL, increases HDL (the “good cholesterol” that removes cholesterol from tissues), and raises nitric oxide levels. It also reduces inflammatory markers like IL-6, TNF-alpha and C-reactive protein, all of which play crucial roles in plaque instability. Regular activity reduces oxidative stress, intensifies antioxidant defenses, and helps the endothelium restore itself more efficiently. Over time, these effects make arteries more flexible, reduces their thickness and stabilize existing plaques, making them less likely to break and cause strokes and heart attack.
According to experimental studies by Laufs et al[4] there is a direct relationship between physical inactivity and vascular dysfunction. The study conducted on mice showed that sedentary animals exhibit remarkably higher levels of vascular oxidative stress compared to those involved in voluntary wheel running. The active mice showed increased level of release of vascular superoxide and lipid peroxidation ultimately leading to high oxidative damage of arterial wall. This study demonstrated that, in genetically modified ApoE-/- mice prone atherosclerosis sedentary behavior markedly accelerated atherosclerotic plaque development[4].
The findings in this study provide strong experimental results that physical activity preserves endothelial integrity and protects vascular health while sedentary behavior enhances oxidative stress, decreases nitric oxide production and promotes atherogenesis.
When body is regularly subjected to movements, blood flow increases boosting shear stresses on vessel walls triggering nitric oxide production. Eventually LDL levels and oxidation of LDL drop together preventing endothelium from damage and slow down plaque buildup and keeps artery healthy. Physical activity is a vascular therapy for our arteries. Four weeks of vigorous exercise training improved coronary endothelial function in patients with asymptomatic coronary atherosclerosis [6].
MANAGEMENT AND TREATMENT
LDL Lowering Therapy
Statins are medications that block activity of enzyme called HMG-CoA reductase. Because of this, liver makes more LDL receptors (LDL-R). These receptors drag LDL-cholesterol out of the blood. So, statins help decrease the amount of LDL-C circulating in blood. They also reduce other harmful lipoproteins. Statins are very efficient and have been proven in many studies to decrease risk of heart diseases[1].
These medicines show their effect in intestine by reducing how much cholesterol your body absorbs from food. This lowers LDL-C. Ezetimibe is usually taken when statins alone are not enough or when a patient who have contraindication to statins[1].
These medicines bind bile acids in intestine so liver has to use more cholesterol to make new bile acids. This lowers LDL-C by 18-25% [1].
Cholestyramine and colestipol generally cause stomach problems and can interact with other drugs. Colesevelam is better accepted and has less interactions, so it can be taken with statins.
Proprotein convertase subtilisin/Kexin type-9 inhibitors are newer medicines. PCSK-9 normally inhibits LDL receptors in the liver.
When PCSK-9 is blocked:
Triglycerides lowering therapy
Statins decrease the plasma concentration of triglyceride-rich lipoproteins by suppressing the enzyme HMG-CoA reductase, important enzyme in the cholesterol biosynthesis. Although hypertriglyceridemia (HTG) is acknowledged as a cardiovascular risk factor, statins remain the first-line treatment because they not only reduce triglycerides but also decrease CV risk. In patients with high triglycerides levels, statins can substantially lower triglycerides-up to about 27%- especially at higher doses.
Fibrates act as peroxisome proliferator-activated receptor-alpha (PPAR-α) agonists. They affect transcription factor that modulate lipid and lipoprotein metabolism, increasing the breakdown and elimination of triglycerides from the blood. Fibrates are particularly efficient in lowering fasting triglyceride levels and improving lipid profiles by decreasing TG-rich lipoproteins and increasing HDL (good cholesterol).
n-3 fatty acids, involving eicosapentaenoic acid (EPA) and docosahexaenoic acid (DHA), are known to reduce TG levels- possibly through interrelations with PPARs that affect lipid metabolism. Although the accurate mechanism is not fully understood, these fatty acids have shown to decrease triglycerides levels by up to 45%. TG levels are not effectively controlled by statins or fibrates alone, n-3 fatty acids can be used in combination therapy. These combinations are generally well-tolerated, effective and safe in achieving further triglyceride reduction[1].
Anti-platelet Therapy
Anti-platelet treatment helps protect patient by reducing the formation of blood clots (thrombus) and lowering inflammation in blood vessel. Aspirin is widely used for this purpose and help prevent more heart issues in people with existing heart or blood vessel disease. Among the four, common treatment used for protecting such conditions (statins, aspirin, ACE inhibitors and beta-blocker), the combination of statins and aspirin has shown the huge decrease in death rate according to studies. Besides aspirin, other anti-platelets drugs like prasugrel, ticagrelor, clopidogrel have also shown good results.
Anti-hypertensive Therapy
Beta-blockers are medication that help decrease the chances of repeated heart attack, sudden heart related death and generally death in people who have had a heart attack. They work by retarding the heart rate and decreasing blood flow pressure which make blood move more smoothly and reduces stress on the artery walls. It has shown that beta-blocker can also lower the progression of atherosclerosis. Another factor involved in this process is angiotensin 2, agent that causes inflammation and rises harmful molecule called ROS (reactive oxygen species). Blocking angiotensin 2 assists decrease inflammation in blood vessels. The medicines that block the renin-angiotensin also help regenerate normal function of the inner lining of blood vessels.
FUTURE PROSPECT
CONCLUSION
Physical activity shows its great impact on cardiovascular health. It helps in handling of cholesterol level, blood vessels functioning and cures diseases like atherosclerosis. Atherosclerosis is a slow, progressive disease where fat, cholesterol and inflammatory cells grow up inside the arteries, this reduces blood flow by narrowing vessels. Inner lining of vessels known as endothelium, plays key role in keeping blood vessels healthy. The stage for atherosclerosis begins when damaging the lining of blood vessels. The endothelium, cholesterol level and proper vascular function is protected by regular physical activity.
REFERENCES







Krutika Mule, Anamika Ghosh, Rounak Titarmare, Mansi Dahalkar, Ayush Khedikar, Ranutai Dharne, Akansha Akre, Interplay Between Sedentary Lifestyle and Atherosclerosis: Insights into Pathogenesis and Therapy, Int. J. of Pharm. Sci., 2026, Vol 4, Issue 4, 514-526. https://doi.org/10.5281/zenodo.19410123
 10.5281/zenodo.19410123
10.5281/zenodo.19410123