
We use cookies to ensure our website works properly and to personalise your experience. Cookies policy
Dayanand Institute of Pharmacy, Latur.
Metachromatic leukodystrophy (MLD) is a rare, inherited lysosomal storage disorder characterized by the progressive demyelination of the central and peripheral nervous systems. It is primarily caused by a deficiency of the enzyme arylsulfatase A (ARSA), leading to the accumulation of sulfatides, which are toxic to myelin-producing cells. MLD presents in various clinical forms—late-infantile, juvenile, and adult—each differing in onset, progression, and severity. Early and accurate diagnosis remains a major challenge due to its clinical heterogeneity and overlap with other neurological conditions. Recent advances in molecular diagnostics, neuroimaging, and biochemical assays have improved early detection and classification of the disease. In parallel, therapeutic research has made significant strides. While hematopoietic stem cell transplantation (HSCT) has shown limited success, newer approaches such as gene therapy, enzyme replacement therapy (ERT), and substrate reduction therapy are currently under investigation, offering hope for more effective disease management. This review provides a comprehensive overview of MLD, detailing its underlying pathogenesis, current diagnostic strategies, and the state-of-the-art in therapeutic interventions.
Metachromatic Leukodystrophy (MLD) is progressive neurodegenerative disorder that affects the white matter of the brain and spinal cord. The abnormal development or destruction of the myelin sheath, the protective covering that insulates nerve cells throughout the central and peripheral nervous system. MLD involves cerebroside sulphate accumulation, like most enzyme deficiencies has an autosomal recessive inheritance pattern.
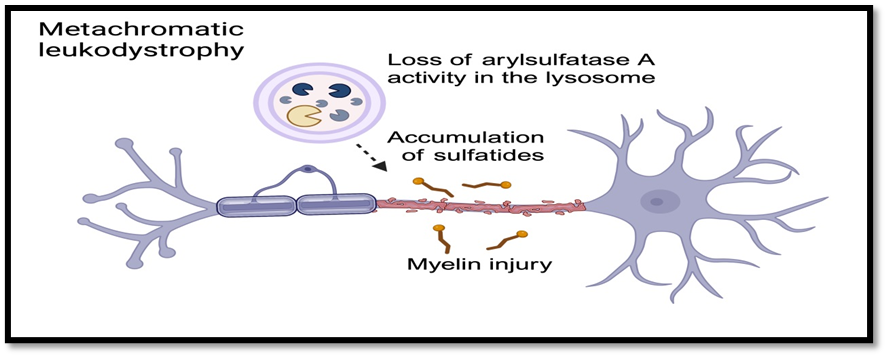
Fig no.1.1 The global Leukodystrophy Initiative
History of Metachromatic Leukodystrophy (MLD)
Metachromatic leukodystrophy has a rich and evolving history in medical science, spanning pathology, biochemistry, and genetics. Here's a detailed look at how the understanding of MLD developed over time:
1. Early Descriptions (1920s–1930s):
2. Recognition as a Distinct Disease (1950s):
3. Biochemical Advances (1960s):
4. Genetic Understanding (1970s–1980s):
5. Subtypes Identified (Late 20th Century):
It involves modifying the patient’s stem cells to express functional ARSA and reintroducing them into the body.
Signs & Symptoms of MLD (metachromatic leukodystropy):
It is characterised into three types
Symptoms:
MLD is caused by a deficiency of the enzyme arylsulfatase A (ARSA), leading to accumulation of sulfatides in the brain and nervous system. This results in progressive demyelination, meaning the protective myelin sheath around nerves breaks down. As a result, nerve signals slow or stop, causing widespread neurological and physical dysfunction.
1. Motor Symptoms (Movement-Related)
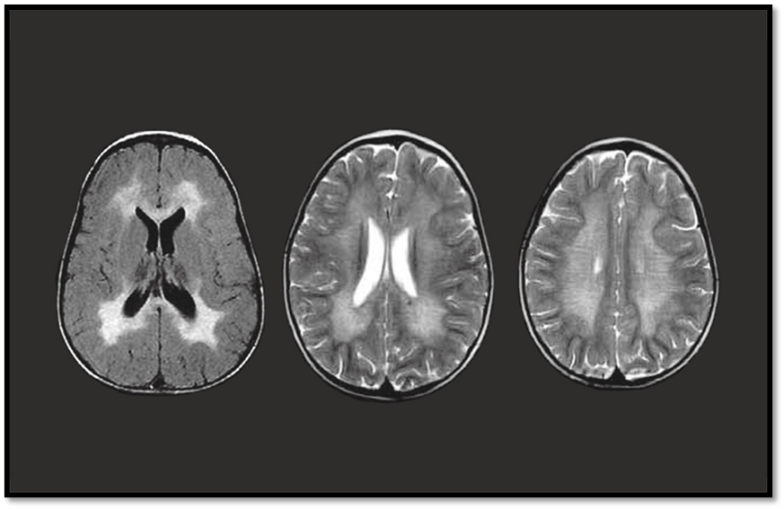
Fig no.1.2 Metachromatic Leukodystrophy
Fig.no.1.3 Spasticity

Fig.no.1.4 Ataxia

Fig.no.1.5.Paralysis

Fig.no.1.6.Tremors
2. Cognitive Symptoms
3. Behavioural and Psychiatric Symptoms
Traditional Formulation:
There is no truly ancient or traditional drug specifically used for Metachromatic Leukodystrophy (MLD), because MLD is a genetic and biochemical disorder only identified in the 20th century.The molecular understanding of MLD (enzyme deficiency, sulfatide accumulation, etc) is a product of modern biomedical science. Ancient medicine systems (like Ayurveda, Traditional Chinese Medicine, or Greco-Arabic medicine) did not have knowledge of lysosomal storage diseases.

Fig no.1.7. traditional formulation
First Therapeutic Attempts (Historical Context)
If you're asking about the earliest treatments in modern medicine, these would include:
3. Enzyme replacement and gene therapy research (2000s-2010s):
Drugs used in treatment of Metachromatic Leukodystrophy
2. Atidarsagene autotemcel
Evolution of MLD:
1. Clinical Presentation: MLD is a rare, autosomal recessive lysosomal storage disease caused by a deficiency in the enzyme arylsulfatase A (ARSA).
Symptoms vary by type:
|
Type |
Onset Age |
Symptoms
|
|
1 Late Infantile |
<4 years |
Developmental regression, gait disturbances, hypotonia, seizures |
|
2 Juvenile |
4-12 years |
Behaviour changes, school difficulties, motor decline |
|
3 Adult |
>16 years |
Psychiatric symptoms, dementia, motor dysfunction |
2. Diagnostic Evaluation
|
Test |
Purpose |
|
MRI Brain |
Shows symmetrical white matter demyelination (especially periventricular regions) |
|
ARSA enzyme assay |
Measures arylsulfatase A activity (low in MLLD) |
|
Urine sulfatides test |
Elevated sulfatides support diagnosis |
|
Generic testing |
Confirms mutations in the ARSA gene |
|
Nerve conduction studies |
May show demyelinating peripheral neuropathy |
|
Neurocognitive assessment |
Baseline for tracking progression |
Treatment:
Hematopoietic Stem Cell Transplantation:
Allogenic hematopoietic stem cell transplantation (HSCT) is considered the standard treatment for presymptomatic and early-symptomatic adult and late-juvenile forms of metachromatic leukodystrophy (MLD), though its benefits in early-onset cases are limited due to rapid disease progression. Following transplantation, patients may initially experience worsening MRI abnormalities and symptoms, likely due to chemotherapy-related neurotoxicity and the delay in full engraftment of donor cells within the brain. However, when performed early in the disease course, HSCT can lead to long-term stabilization or even improvement in white matter changes. Traditionally, the therapeutic effect of HSCT was believed to occur through cross-correction, where donor-derived macrophages and microglia provide functional arylsulfatase A (ASA) to the patient’s deficient neural cells. This mechanism, however, has been called into question, as studies have shown that ASA secreted by these donor cells often lacks the mannose-6-phosphate necessary for cellular uptake. Furthermore, postmortem analysis of HSCT-treated MLD patients revealed ASA presence only in donor macrophages—not in oligodendrocytes or astrocytes—suggesting that cross-correction may play a minimal role. Instead, an alternative mechanism has been proposed in which donor macrophages contribute metabolically by clearing sulfatide buildup and exerting anti-inflammatory effects that promote oligodendrocyte survival and remyelination. This is supported by findings of higher oligodendrocyte counts and signs of remyelination in transplanted patients. Similar observations have been made in HSCT-treated mouse models of Krabbe disease, where benefits occurred despite limited cross-correction, further highlighting the role of donor-derived immune cells in reducing neuroinflammation. Despite these therapeutic potentials, HSCT carries significant risks, including graft-versus-host disease, infections, toxicity, chronic rejection, and increased malignancy risk, with mortality rates around 10–15%, and even higher in some patient groups.
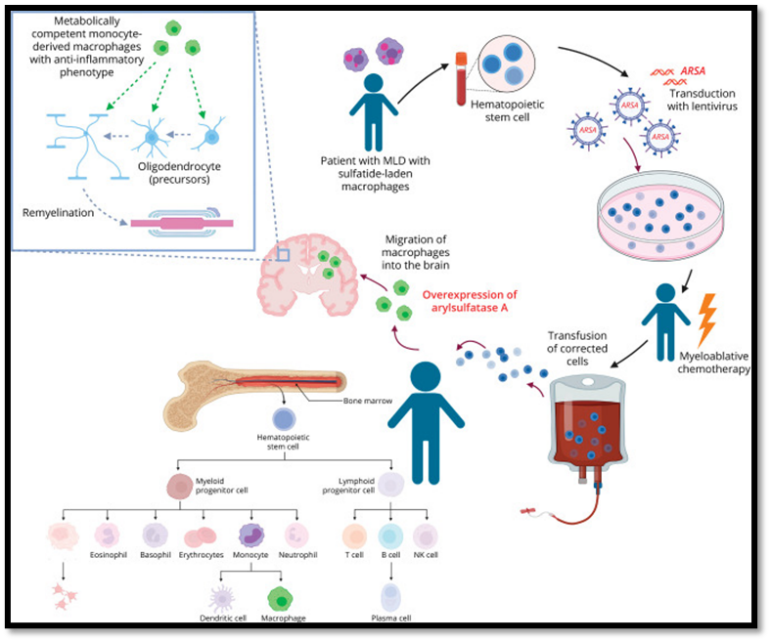
Fig no.1.8 Hematopoietic Stem Cell Transplantation
Intracerebral Gene Therapy:
One of the disadvantages of HSCT and HSC-GT is the delayed delivery of ASA-expressing cells. Therefore, more efficient delivery methods are needed, also able to target glial cells and neurons. Intracerebral injections of a viral vector (AAVrh.10-hARSA) have been tested in nonhuman primates and were safe and effective. In a phase I-II clinical trial, 3 patients with late-infantile MLD (2 presymptomatic and 1 early-symptomatic) and 1 early-juvenile early-symptomatic patient, aged 9 months to 5 years, were treated with this vector. AAVrh10-hARSA was detected in urine, and significant increase in ASA activity was observed in the CSF. However, all patients had clinical and radiologic disease progression similar to or more rapid than the natural history of MLD. T2 hyperintense areas developed around the injection sites on brain MRI. Why this approach failed in humans is not known. Of interest, similar MRI changes were seen in participants in an intracerebral gene therapy trial for Sanfilippo disease, shown to be due to extracellular spilling of lysosomal enzymes with subsequent WM damage.
Transplantation:
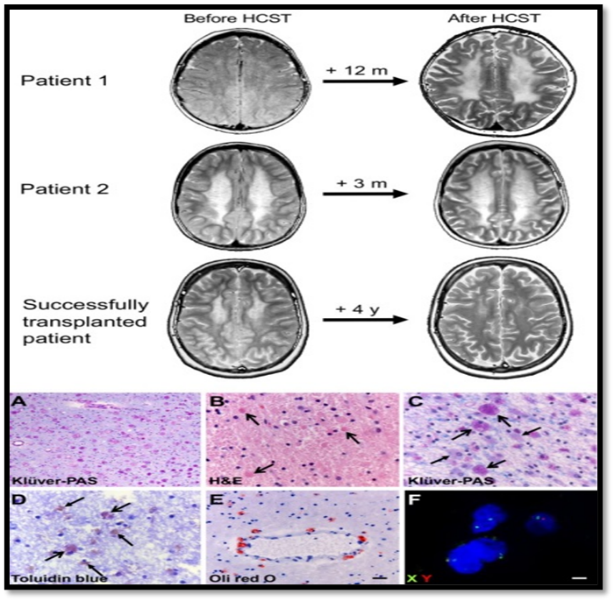
Fig no.1.4 Effects of Transplantation
Effects of transplantation in MLD.:
Part 1: Evolution of brain MRI in patients 1 and 2 and, as an example, evolution of white matter abnormalities in a patient successfully transplanted (axial T2?weighted images shown). In the two deceased patients, white matter abnormalities increase, especially in patient 1. The successfully transplanted patient illustrates improvement of leukodystrophic changes 4 years after HSCT. Part 2: Donor cells reach the brain of transplanted MLD patients. (A) Stain with Klüver (blue dye for myelin) and periodic acid Schiff (PAS, pink, stain for sulfatides in macrophages) of the cerebral periventricular white matter of an untreated MLD patient (patient 5) shows loss of myelin and abundance of cells loaded with PAS?positive granular material. (B) Hematoxylin & Eosin stain of the frontal subcortical white matter of a HSCT?treated patient (patient 1) shows the presence of macrophages with intense eosinophilic cytoplasm (arrows) next to macrophages loaded with clearer granular material. (C) A Klüver?PAS stain of the same region of this patient confirms the presence of a double population of macrophages, more (open arrows) and less (closed arrows) intensely PAS positive. (D) Toluidine blue stain of the parietal white matter of this HSCT?treated patient (patient 2) reveals that only a subset of macrophages is metachromatic (purple, i.e., loaded with sulfatides), the remaining being orthochromatic (brown) and as such able to degrade sulfatides. (E) Metabolic competence of a subset of macrophages in the white matter of a HSCT?treated patient (patient 1) is confirmed by their ability to digest sulfatides, as shown in this Oil Red O stain for neutral fats. (F) In this patient, FISH against the X and Y chromosomes confirms cells of both sexes.
Enzyme Replacement Therapy:
IV enzyme replacement therapy (ERT) stabilizes or improves non-CNS symptoms of lysosomal storage diseases. MLD mouse model studies demonstrated improvements in motor and behavioral symptoms, prompting further investigation in a clinical trial. To assess the safety and efficacy of IV recombinant human (rh)ASA, dose-escalated IV rhASA was administered every 14 days for 52 weeks to 13 patients with MLD with an onset ≤4 years. No serious adverse events related to the treatment were reported. While peripheral nerve pathology did not worsen, motor and cognitive functioning continued to deteriorate, suggesting that IV rhASA does not cross the blood-brain barrier in therapeutic quantities. Nonetheless, the results concerning peripheral nerves indicate that rhASA may have some positive effects on patients with MLD, perhaps due to better penetration through the (homeostatic) blood-nerve barrier, which is leakier than the blood-brain barrier.
Intrathecal administration of rhASA was, therefore, evaluated as an alternative to IV ERT in a clinical trial involving 24 children with MLD who had an onset ≤30 months conducted over 38 weeks. Different dosages ranging from 10 mg to 100 mg were tested across several cohorts. No serious adverse events related to rhASA were reported, although 25% of patients experienced adverse events associated with the intrathecal device or drug delivery method. An overall decline in motor function was observed over time, but patients receiving the highest dose showed the least pronounced decline. The treatment was well tolerated, leading to an extension to evaluate long-term safet). In addition, an ongoing phase 2b trial investigated the effects of a higher dose of 150 mg weekly of rhASA in 36 patients with MLD. This dose corrected biochemical defects, and delayed pathologic features seemed to delay neurologic deterioration and structural changes in some children However, a lack of efficacy on the primary end point (gross motor function) led to discontinuation of this treatment strategy.
REFERENCES





Sachitanand Biradar, Harshvardhan Shinde, Nikhil Chapule, Govind Soni, Vishnu Bharti, Metachromatic Leukodystrophy: A Comprehensive Review of Pathogenesis, Diagnosis, and Therapeutic Advances, Int. J. of Pharm. Sci., 2025, Vol 3, Issue 8, 2828-2839. https://doi.org/10.5281/zenodo.16950440
 10.5281/zenodo.16950440
10.5281/zenodo.16950440