Womens College of pharmacy, Peth Vadgaon.
Myositis refers to a group of rare inflammatory muscle disorders characterized by muscle weakness, fatigue, and chronic inflammation. The major types include polymyositis (PM), dermatomyositis (DM), inclusion body myositis (IBM), and necrotizing autoimmune myopathy (NAM). The exact etiology remains unclear, but genetic predisposition, autoimmune mechanisms, and environmental triggers, including infections and certain medications, are implicated. Diagnosis is based on clinical features, muscle enzyme levels, electromyography, MRI, and muscle biopsy findings. Autoantibodies, such as anti-Jo-1 and anti-Mi-2, play a crucial role in differentiating subtypes. Treatment primarily involves corticosteroids, immunosuppressant, and, in refractory cases, biologic therapies like Rituximab or intravenous immunoglobulin (IVIG). Physical therapy is essential to maintain muscle function. Recent research focuses on novel biomarkers, targeted therapies, and the role of immune modulation. Despitead vancements, disease prognosis varies with IBM being particularly resistant to treatment. Future studies are needed to explore more effective and personalized therapeutic approaches. This review highlights the pathophysiology, clinical features, diagnosis, and evolving treatment strategies for myositis.
In addition to being referred to as myositis, idiopathic inflammatory myopathies consist of the following conditions: dermatomyositis (DM), polymyositis (PM), necrotizing myopathy (NM), and inclusion body myositis. Every single one of the msuffers from muscle weakness. In order to arrive at a diagnosis, the clinical examination, which includes the distribution of paresis, is supplemented with the findings of laboratory tests such as creatine kinase (CK), autoantibodies, electromyography (EMG), and skeletal muscle histology. When it comes to choosing acceptable muscles for biopsy and recognizing patterns of injured muscles that go beyond clinical presentation, magnetic resonance imaging (MRI) of the skeletal muscle can be quite helpful. Because of this, muscle dystrophies can be ruled out.[1] Histopathological and immunochemical analysis of inflammatory muscular infiltrates has improved the diagnosis and the discovery of several new auto-antibodies leading to the identification of more homogeneous subsets of myositis. Notably, the increasing knowledge has been translated into new therapeutic targets in patients affected with IIM. However, several issues remain unresolved, including the absence of universally accepted guidelines for the treatment of each subset of disease, the limited number of randomized controlled trials for most of the drugs used in patients with IIM and the limited efficacy of the immunosuppressive and immunomodulatory drugs currently available in patients with inclusion body myositis (IBM) On the other hand, toxic compounds, such as those utilized in rheumatologic and IIM treatments, have the potential to cause damage to the muscles. During the process of diagnosing muscle disease, It is essential to take into consideration drug-induced myopathies, which might be difficult for medical professionals to identify.[2]Asymmetric muscle involvement is characterized by a weakening that is often more severe in the arm and leg that is not the dominant one.[3] Resistant organ inflammation is a pathomechanism that is involved in the majority of immune-mediated illnesses. [4]
Etiology:
Dr. Frederick W. Miller (FDA): Idiopathic inflammatory myopathies are defined as having unknown etiology. Recent research suggests that environmental factors, specifically Gm3; 5, may affect genetically plicate organisms. [5] Environmental factors may contribute to systemic connective tissue diseases, as evidenced by the sudden development of myositis as a viral-like illness in many cases. Recent data suggests that myositis develops in non-random patterns, with clusters of new cases recorded in India. [6] and North America [7] Immunologic variables are most likely implicated in the pathology of the disease. Dermatomyositis is thought to because biogenetic factors, while some investigations have found no link between HL a haplotype and illness. [8] Both viral and noninfectious factors may cause myositis. Picorna viruses, an infectious environmental agent that can cause myositis, have indirect support from serologic, ultra structural and animal model studies. The known causes are diverse, including medicines, poisons, and some infectious agents; however, in the majority of instances, a cause has yet to be found.[9] Retroviruses, including HIV and T- lympho trophic virus type 1, are important.[10]
Pathogenesis:
Auto anti-bodies have astrong correlation with illness symptoms. As a result, auto antibodies provide valuable information for clinical diagnosis, classification, prognosis prediction, and therapy decision- making in IIM patients. Over the past decade, new myositis-specific auto antibodies have been discovered. [1, 11] Diabetes affects 1·4 and 5·8 individuals per100, 000 in the US, respectively. The study reveals safe male predominance and increased incidence among elderly individuals. Juvenile Diabetes Mellitus (JDM) affects3.2million youngsters in the UK and is more pre valentingirls. [12] DM pathogenesis involves immune complexes attaching to endothelial cells, activating complement system & causing cell lysis via the membrane- attack complex (MAC). This causes cell death and fewer capillaries in the muscle. [13] We have limited knowledge about epidemiology. In small cohortstudies, 61% of the participants were male. Dysphasia, a prevalent symptom of IBM, is often overlooked and under-reported as a presenting symptom (42,116). However, it can be identified through a history or radiological investigations (116-117). As the disease advances, dysphasia can lead to nutritional deficiencies, weight loss, and aspiration pneumonia, a leading cause of mortality in IBM. [14, 17]
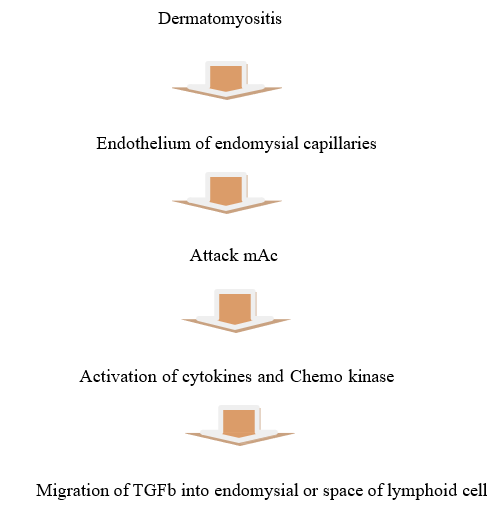
Fig1: Pathogenesis Of Myositis
Physical Features
Physical examination findings are so valuable that muscle histology may not be necessary for diagnosing IBM.
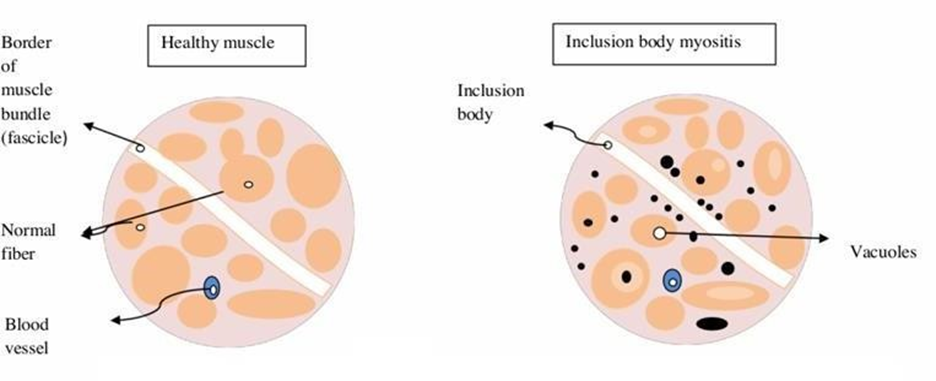
Fig2: Physical Features
IBM muscle fibers are characterized by the presence of clusters of cellular material known as inclusion bodies as well as empty spaces that resemble bubbles.[16] There is a significant rate of incorrect diagnosis as a result of the fact that the physical examination signs of IBM are not frequently recognized. (17) The progression of the disease can result in dysphasia, which is characterized by difficulty swallowing [18]. In spite of the fact that IBM is not frequently reported as a presenting symptom, it is frequently discovered by history or radiological testing.[19] Either quickly or gradually over the course of time, diabetes can develop. The most prevalent symptom of muscle weakness is weakness in the muscles, which usually affects the proximal muscles. Myalgias are a less common symptom of muscle weakness. The proximal muscles typically experience a reduction in strength as a consequence of contractures. Muscular atrophy, which accounts for forty percent of all cases, manifests itself later in the development of the disease. When the condition is severe, involvement of respiratory & or pharyngeal muscles can lead to dysphasia, breathing difficulties, and a biggest is pneumonia. [20]
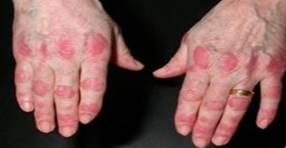
A. Gattron’s papules
Fig.3 symptoms a. Gattron’s papules
Papules with a violaceous tint that are located over the dorsal-lateral aspect of interphalangeal and/or metacarpophalangeal joints are referred to as A. Gattron's papules. When they are fully developed, these papules develop a little depression in the centre, which might give them a white and atrophic appearance. There is a possibility of association with telangiectasia. Poikilodermatomyositis, also known as circumscribed violaceous erythema [20].
Diagnosis: A reliable diagnosis of myositis requires a detailed medical history and examination.
[1] A biopsy is necessary to accurately diagnose myositis and distinguish between its various subtypes. It's important to rule out muscular dystrophy or other inherited myopathies. To achieve this, choose a representative muscle for the biopsy, often with significant paresis. MRI can detect final stages of tissue break down. [1] For the purpose of this study, a simplified taxonomy of terms in the field is utilized, as demonstrated below: Creating intelligent machines that are able to think and interact with their surroundings is the primary emphasis of the field of computer science known as artificial intelligence (AI).The myositis- specific auto antibody is an important diagnostic tool, and there are a number of different methods that can be used to detect myositis-specific auto antibodies or MAAs. Each of these methods has its own set of benefits and drawbacks, including sensitivity, specificity, throughput, cost, and the level of expertise that is required for detection. In common diagnostic laboratories, the Testing procedures that are considered standard include gel-based counter-immune techniques and indirect immunofluorescence with HEp2 cells. In addition to enzyme-linked immunosorbent assays, electrophoresis and immune diffusion are also included in the techniques under consideration. [21-22] ILDs can be evaluated with a chest X-ray. Patients who have anti-CADM and anti- synthase autoantibodies are candidates for high-resolution computed tomography (CT) imaging. With this method, lung changes can be identified before clinical manifestations appear.[23]
Treatment:
Corticosteroids
In the therapy of inflammatory myopathy, corticosteroids continue to be the medication of choice for the initial phase. The severity of the muscle issue is taken into consideration when determining the starting dose. The standard treatment for myositis that has recently manifested itself or for a significant flare-up is a dose of 1mg/kg, which is equivalent to60 to 80mg/day, separated into two or three doses. In order to restore muscle enzymes to normal levels, continue for four to eight weeks. Over a period of six months, the dose is gradually decreased to between five and ten milligrams per day when responsiveness and adverse effects are taken into consideration. You will continue to take the maintenance dose of 5-10 mg/d for another six months, or until you become active.[24] Rituximab: B cell depletion Rituximab is the most promising biological therapy thus far. B-cell blockage is based on the presence of auto antibodies, which indicate that B cells have a role in myositis. Up to Our unpublished research suggests that 90% of myositis patients may have auto antibodies, with B and plasma cells seen in muscle tissue. The' Rituximab in Myositis'(RIM) experiment was based on case series data collected between 2005 and 2012. [25]
Calcineurin inhibitors
Calcineurin inhibitors may be a alternative choice if standard therapy fails to provide enough results. Other immunosuppressive medications. Although Calcineurin inhibitors show promise in treating myositis-associated ILD, more randomized prospective multicenter studies are needed. [26]
Diet and lifestyle
There is minimal data on dietary interventions in IIM patients. A6-month Randomized controlled experiment found that combining creatine and exercise improved muscle performance in adults compared to placebo. Creatine supplementation was not related with any adverse events; nevertheless, attention should be exercised when treating patients with kidney impairment. [27]
CONCLUSION
Myositis encompasses a diverse group of inflammatory muscle diseases with complex and multifactorial pathogenesis involving immune dysregulation, genetic susceptibility, and environmental triggers. Advances in our understanding of autoantibodies, cytokine signaling, and cellular mechanisms have enhanced diagnostic accuracy and the classification of myositis subtypes. Despite progress, early and accurate diagnosis remains challenging due to overlapping symptoms with other neuromuscular disorders. Emerging imaging techniques, autoantibody profiling, and muscle biopsy remain essential tools in the diagnostic process. Treatment has traditionally relied on immunosuppressive therapy, particularly corticosteroids and DMARDs, but novel biologics and targeted therapies are showing promise in refractory cases. Continued research is critical to unravel disease-specific mechanisms and to develop personalized, effective, and less toxic therapeutic strategies. Past research on myositis provided significant insights into the clinical and pathological diversity of this group of idiopathic inflammatory myopathies. Over the years, various subtypes—including polymyositis, dermatomyositis, and inclusion body myositis—were recognized, each with distinct clinical features, histopathological findings, and responses to therapy. Investigators had increasingly emphasized the role of autoimmune mechanisms in disease pathogenesis, supported by the discovery of myositis-specific and myositis-associated autoantibodies. These findings contributed to improved classification criteria and diagnostic accuracy. Advancements in imaging techniques such as MRI, along with the use of muscle biopsy and serological markers, enhanced the early detection and monitoring of disease activity. Despite these developments, treatment remained challenging, particularly in refractory cases and in subtypes such as inclusion body myositis, which responded poorly to conventional immunosuppressive therapies. Earlier studies often relied on corticosteroids and broad-spectrum immunosuppressants, though long-term outcomes varied widely among patients. Overall, the literature underscored the importance of individualized treatment approaches and highlighted the limitations of existing therapies. It also pointed to the need for further exploration into the molecular and immunological underpinnings of myositis. As a result, the field increasingly turned toward targeted therapies and biologics, guided by emerging biomarkers and mechanistic studies. These foundational studies laid the groundwork for current and future research aimed at optimizing patient outcomes and understanding the complex nature of myositis. In the future, the understanding and management of myositis are expected to evolve significantly through advances in immunology, genetics, and personalized medicine. As research continues to unravel the molecular and cellular mechanisms driving the different subtypes of myositis, more precise classification systems and diagnostic tools are likely to emerge. The identification of novel autoantibodies, genetic markers, and molecular signatures will enhance early diagnosis, disease stratification, and prediction of treatment response. Therapeutically, future approaches will likely shift toward targeted and individualized treatment strategies. Biologic agents and small molecule inhibitors, guided by a patient's specific immunologic profile, may replace or complement traditional immunosuppressive therapies. Additionally, regenerative medicine and gene-based therapies hold potential in restoring muscle function and slowing disease progression, particularly in forms like inclusion body myositis, which have shown resistance to standard treatments. Artificial intelligence and machine learning may further support clinical decision-making by integrating clinical, serologic, imaging, and genetic data to tailor management plans. Moreover, global collaboration and patient registries will likely enhance the quality and scale of clinical trials, enabling better evaluation of emerging therapies. In summary, the future of myositis research and treatment lies in a more precise, mechanism-based approach, driven by technological and scientific innovation. These advancements hold promise not only for improving outcomes but also for enhancing the quality of life for individuals living with this complex group of inflammatory muscle diseases.
REFERENCES








Nandrekar A. N.*, Patil Y. N., Palake G. J., Patil S. K., Jadhav V. S., Nalavade P. A., M. S. Patil, Dr. D. R. Jadage, Myositis: A Comprehensive Review of Pathogenesis, Diagnosis, And Treatment, Int. J. of Pharm. Sci., 2025, Vol 3, Issue 4, 2972-2979 https://doi.org/10.5281/zenodo.15275091
 10.5281/zenodo.15275091
10.5281/zenodo.15275091
