
We use cookies to ensure our website works properly and to personalise your experience. Cookies policy
Shiva Trust's Godavari College of Pharmacy, Manori, Nashik – 422004, Maharashtra, India.
Alzheimer’s disease (AD) is a progressive neurodegenerative disorder marked by memory loss, cognitive decline, and behavioral changes. The disease develops through several mechanisms, including amyloid-beta plaque formation, tau protein hyperphosphorylation, oxidative stress, inflammation, and synaptic failure. Neuropharmacological treatments aim to influence these processes to relieve symptoms and slow disease progression. Current drugs mainly include cholinesterase inhibitors, which strengthen cholinergic activity, and NMDA receptor antagonists, which balance glutamatergic transmission. New therapeutic directions focus on disease modification by targeting amyloid and tau pathology, oxidative stress, and neuroinflammatory pathways. Advances in molecular neuropharmacology — such as monoclonal antibodies, small-molecule inhibitors, and neuroprotective compounds — may lead to more effective therapies. Understanding the complex molecular and neurochemical basis of AD is essential for developing next-generation drugs to enhance cognitive health and quality of life.
Alzheimer’s disease (AD) is a chronic and progressive neurodegenerative condition that represents the most common cause of dementia, leading to gradual deterioration in memory, cognition, and behavioral functions (1). The disease is characterized by two major pathological signatures: the build-up of amyloid-β (Aβ) deposits outside neurons and the formation of neurofibrillary tangles inside neurons due to abnormal phosphorylation of tau protein (2,3). These abnormalities impair synaptic communication and ultimately result in neuronal degeneration. AD pathology also includes widespread neurochemical imbalances, such as cholinergic deficits, glutamate-induced excitotoxicity, increased oxidative stress, mitochondrial instability, and persistent neuroinflammatory responses, all of which collectively contribute to cognitive decline (4,5).
Current treatments primarily address symptoms rather than modifying the disease course. Drugs like acetylcholinesterase inhibitors aim to improve cholinergic transmission, while N-methyl-D-aspartate (NMDA) receptor antagonists help regulate excessive glutamatergic activity; however, their ability to slow progression remains limited (6). Newer neuropharmacological strategies focus on altering underlying mechanisms, including inhibition of Aβ and tau pathology, modulation of inflammatory processes, reduction of oxidative damage, and preservation of synaptic function, offering potential for disease modification (7,8). A deeper understanding of these molecular pathways is essential for developing more effective therapies that can enhance cognitive outcomes and overall quality of life for individuals with AD.
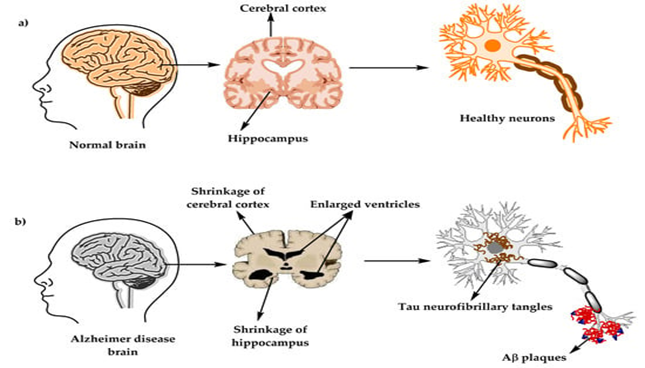
Figure.01
PATHOPHYSIOLOGY :
Alzheimer’s disease (AD) is a chronic, progressive neurodegenerative condition that primarily affects the hippocampus and cerebral cortex, resulting in progressive cognitive decline, memory impairment, and behavioral alterations. The pathogenesis of AD involves several interconnected molecular pathways that ultimately cause neuronal loss and synaptic dysfunction.
1. Amyloid Cascade Mechanism
A major pathological hallmark of AD is the deposition of β-amyloid (Aβ) peptides in the brain. These peptides are produced when amyloid precursor protein (APP) is abnormally cleaved by β-secretase and γ-secretase enzymes. The accumulated Aβ forms insoluble plaques that trigger neuroinflammatory responses, oxidative injury, and synaptic toxicity. Activation of microglial cells around plaques leads to the release of inflammatory cytokines and neurotoxic molecules, worsening neuronal damage (7,8).
2. Tau Protein Abnormalities
Another crucial mechanism involves tau protein hyperphosphorylation and aggregation within neurons. Normally, tau maintains microtubule stability, but when excessively phosphorylated, it detaches and forms neurofibrillary tangles (NFTs). This disrupts axonal transport and intracellular communication, leading to neuronal degeneration. The propagation of tau pathology throughout brain regions closely parallels the clinical progression of cognitive deficits (9).
3. Neuroinflammation and Oxidative Stress
Chronic activation of glial cells contributes to persistent neuroinflammation. Reactive oxygen species (ROS) and nitric oxide generated during this process damage cellular proteins, DNA, and lipids. Oxidative stress also enhances Aβ deposition and tau phosphorylation, creating a self-perpetuating cycle of neuronal dysfunction and death (10).
4. Cholinergic System Degeneration
Loss of cholinergic neurons in the basal forebrain is an early and characteristic event in AD pathology. The reduction in acetylcholine synthesis and release impairs learning and memory processes. This neurotransmitter deficit forms the pharmacological basis for the use of acetylcholinesterase inhibitors, which aim to improve cognitive symptoms by increasing synaptic acetylcholine availability (11).
5. Mitochondrial and Synaptic Dysfunction
Mitochondrial impairment results in decreased ATP generation and excessive oxidative stress, compromising neuronal metabolism and synaptic transmission. Synaptic loss, rather than plaque or tangle density, correlates most strongly with cognitive deterioration. Mitochondrial dysfunction therefore plays a pivotal role in amplifying neuronal vulnerability in regions with high energy demand, such as the hippocampus (12)
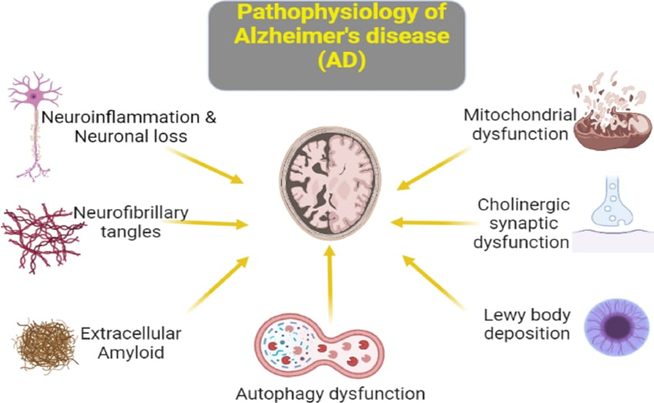
Figure :02
Neurobiology of Alzheimer disease :
Alzheimer’s disease (AD) involves complex molecular and cellular changes that impair cognitive and behavioral function. Two key pathological features are extracellular amyloid-beta (Aβ) plaques and intracellular neurofibrillary tangles (NFTs) composed of hyperphosphorylated tau proteins (8,14). Aβ accumulation triggers oxidative stress, inflammation, and synaptic dysfunction, while tau pathology leads to microtubule instability and neuronal death.
Cholinergic deficits in the basal forebrain contribute to memory loss, whereas glutamatergic excitotoxicity via NMDA receptor overactivation causes neuronal injury (15,5). Mitochondrial dysfunction, defective autophagy, and altered insulin signaling further worsen neuronal degeneration (17). Neuroinflammatory activation of microglia and astrocytes releases proinflammatory cytokines, amplifying damage (18). Oxidative stress, metal imbalance, and vascular dysfunction also play key roles in disease progression (19,20).
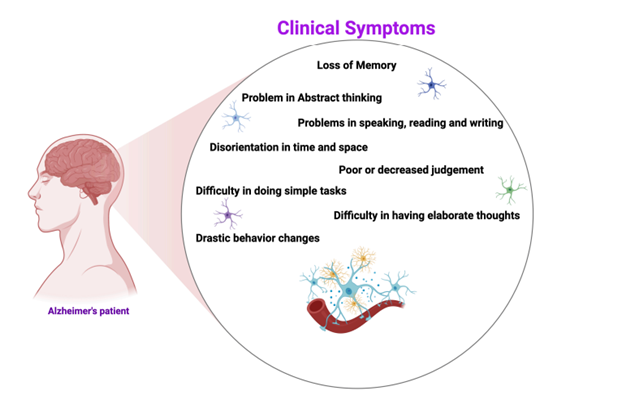
Figure:03
Role of various neurotransmitters and their receptors in Alzheimer’s disease :
New therapeutic stratergies for the treatment of Alzheimer disease :
Current therapies mainly relieve symptoms but have limited impact on disease progression. Emerging treatments target disease mechanisms directly.
CONCLUSION :
Alzheimer’s disease is a complex and progressive neurodegenerative condition marked by continual deterioration in cognition and widespread neuronal damage. Its neuropharmacological basis involves multiple interconnected mechanisms, including disturbances in neurotransmitter pathways, accumulation of amyloid-β peptides, abnormal phosphorylation of tau protein, oxidative imbalance, and persistent neuroinflammation. Although significant progress has been made in understanding these mechanisms, currently available medications primarily alleviate symptoms and do not substantially slow disease progression. Newer therapeutic approaches—such as neuroprotective agents, receptor-modulating compounds, inhibitors targeting amyloid and tau abnormalities, and anti-inflammatory interventions—show encouraging potential for modifying disease outcomes. Advancing molecular research, along with genetic discoveries and innovative drug-delivery technologies, will be crucial for developing more effective, disease-modifying treatments for Alzheimer’s disease in the coming years.
ACKNOWLEDGEMENTS :
Preparing this review on the neuropharmacology of Alzheimer’s disease has been an enriching and insightful experience. I am grateful to my mentors and colleagues for their constant guidance and encouragement throughout this work. I also appreciate the contributions of all researchers whose studies formed the foundation of this review.
REFERENCE


Bhumika Satdive, Shweta Padosa, Yashodeep Patil, Neuropharmacology of Alzheimer Disease, Int. J. of Pharm. Sci., 2025, Vol 3, Issue 11, 3989-3994. https://doi.org/10.5281/zenodo.17711757
 10.5281/zenodo.17711757
10.5281/zenodo.17711757