
We use cookies to ensure our website works properly and to personalise your experience. Cookies policy
Shiva Trust’s Godavari College of Pharmacy, Manori, Nashik - 422004, Maharashtra, India
Quality by Design (QbD) is a regulatory-endorsed, systematic, and science-based approach that emphasizes comprehensive process understanding and risk-based control strategies to ensure consistent pharmaceutical product quality. Unlike conventional development paradigms that rely heavily on end-product testing, QbD embeds quality within formulation and process design from the earliest stages of development. This systematic review critically evaluates the application of QbD principles in pharmaceutical formulation development and assesses their influence on product quality, process robustness, manufacturing efficiency, and regulatory compliance throughout the product lifecycle. A comprehensive literature search was conducted in PubMed, Google Scholar, ScienceDirect, SpringerLink, and Wiley Online Library in accordance with PRISMA guidelines. Peer-reviewed articles published between 2010 and 2025 that reported the implementation of QbD tools in formulation and process development were included. The review highlights the extensive use of core QbD elements, including Quality Target Product Profile (QTPP), identification of Critical Quality Attributes (CQAs), risk assessment methodologies, Design of Experiments (DoE), and Process Analytical Technology (PAT), across diverse dosage forms such as solid oral dosage forms, modified-release systems, nanoformulations, and advanced drug delivery platforms. The findings demonstrate that QbD-based development enhances formulation robustness, minimizes batch-to-batch variability, improves process understanding, and supports regulatory flexibility through the establishment of design space and risk-based control strategies. Despite challenges associated with increased initial resource requirements, data complexity, and the need for specialized expertise, QbD provides a robust framework for continuous improvement, efficient scale-up, and effective lifecycle management. Overall, the adoption of QbD aligns with global regulatory expectations and represents a critical shift toward more reliable, efficient, and patient-centric pharmaceutical development. Among the included studies, approximately 70–80% employed Design of Experiments (DoE) as a core QbD tool, while 45–60% reported establishment of design space. Advanced tools such as Process Analytical Technology and Real-Time Release Testing were applied in nearly 20–30% of studies, reflecting gradual industrial adoption. Furthermore, the review highlights the growing relevance of QbD in ANDA and NDA submissions, where risk-based development supports regulatory flexibility and lifecycle management.
Pharmaceutical formulation development has historically relied on empirical and trial-and-error methodologies, often leading to inconsistent product quality, limited process understanding, and challenges during scale-up and technology transfer. Such conventional approaches frequently fail to account for the complex interactions between formulation variables and process parameters, resulting in variability in critical quality attributes and inefficient manufacturing practices.
The increasing complexity of pharmaceutical dosage forms, including modified-release systems, nano-enabled drug delivery platforms, and biopharmaceutical products, coupled with heightened regulatory scrutiny, has necessitated a paradigm shift toward more systematic and science-based development strategies. In this context, ensuring consistent product performance and patient safety throughout the product lifecycle has become a central focus of regulatory agencies worldwide.
Quality by Design (QbD), as defined in the International Council for Harmonisation (ICH) guideline Q8(R2), is a systematic, science-based approach that begins with predefined objectives and emphasizes thorough product and process understanding, quality risk management, and continual improvement. QbD integrates quality into formulation and process design rather than relying solely on end-product testing, thereby promoting a proactive approach to quality assurance.
Regulatory authorities such as the United States Food and Drug Administration (US FDA) and the European Medicines Agency (EMA) actively encourage the adoption of QbD principles to enhance manufacturing robustness, facilitate regulatory flexibility, and support lifecycle management. The QbD framework is further reinforced through complementary ICH guidelines, including Q9 (Quality Risk Management), Q10 (Pharmaceutical Quality System), and Q11 (Development and Manufacture of Drug Substances), which collectively provide a harmonized foundation for modern pharmaceutical development.
Although QbD has been widely applied across various formulation and process development studies, its implementation remains inconsistent across the pharmaceutical industry. Furthermore, existing literature often addresses QbD applications in a fragmented manner, focusing on specific dosage forms, analytical method development, or regulatory perspectives in isolation. A comprehensive synthesis that integrates formulation development, risk-based tools, regulatory expectations, and emerging technological advancements remains limited.
Beyond scientific and regulatory advantages, Quality by Design offers significant economic and business benefits to pharmaceutical manufacturers. QbD-based development has been associated with a reduction in the cost of poor quality, including fewer batch failures, lower rejection rates, and decreased rework. Systematic process understanding enables early identification of critical risks, thereby minimizing late-stage development failures. In addition, QbD facilitates shorter development timelines and faster time-to-market by reducing experimental redundancy and enabling smoother scale-up and technology transfer. These economic advantages make QbD a strategic tool for improving long-term manufacturing efficiency and competitiveness.
Need for the Review
Despite the growing body of research demonstrating the benefits of QbD in pharmaceutical formulation development, there is a lack of consolidated and systematic evaluation of its practical implementation, commonly employed tools, regulatory relevance, and associated challenges. This systematic review aims to bridge this gap by critically analyzing published literature on QbD-based formulation development, with an emphasis on its impact on product quality, process robustness, and regulatory compliance. By integrating scientific, regulatory, and technological perspectives, this review provides a comprehensive reference for researchers, formulation scientists, and regulatory professionals.
Unlike previous reviews that primarily focus on individual QbD tools such as Design of Experiments or analytical method development, the present review provides an integrated perspective encompassing formulation science, regulatory expectations, real-time release testing, and emerging Industry 4.0 concepts. This review uniquely synthesizes experimental evidence with regulatory science, offering a lifecycle-oriented evaluation of QbD implementation across diverse dosage forms and manufacturing paradigms.
Growth in QbD-Related Publications (2010–2025)
Over the past decade and a half, there has been a marked and sustained increase in scientific publications addressing Quality by Design in pharmaceutical development. Literature indexed in major databases such as PubMed, Scopus, and ScienceDirect demonstrates a steady rise in QbD-focused research beginning around 2010, coinciding with broader regulatory adoption of ICH Q8(R2), Q9, and Q10 guidelines. Early publications primarily emphasized conceptual frameworks and proof-of-concept studies, whereas more recent literature increasingly reports experimental formulation development, case studies, and industrial-scale applications.
Between 2015 and 2025, QbD-related publications expanded significantly to include complex dosage forms, nanoformulations, biologics, continuous manufacturing, and real-time release testing. This trend reflects a transition from theoretical acceptance of QbD toward its practical implementation as a mainstream development strategy. The growing volume and diversity of publications highlight increasing academic and industrial engagement with QbD as a critical component of modern pharmaceutical science.
A bibliometric assessment using PubMed and Scopus databases indicates a pronounced growth in QbD-related publications over the last 15 years. Fewer than 50 publications per year were reported prior to 2012, whereas annual publications exceeded 250–300 articles by 2023–2024. This exponential increase reflects the transition of QbD from a conceptual regulatory framework to a routinely applied development strategy in both academia and industry.

Fig.1. Year vs number of QbD publications
Increasing QbD Expectations in ANDA and NDA Filings
Regulatory expectations for the application of Quality by Design principles in New Drug Applications (NDAs) and Abbreviated New Drug Applications (ANDAs) have progressively strengthened over the last decade. Regulatory agencies, particularly the US Food and Drug Administration and the European Medicines Agency, increasingly encourage applicants to demonstrate systematic product and process understanding through well-defined Quality Target Product Profiles, identification of Critical Quality Attributes, risk-based justification of Critical Process Parameters, and scientifically established design space.
Recent regulatory reviews and guidance documents indicate that submissions incorporating QbD elements benefit from enhanced regulatory confidence, improved assessment efficiency, and greater post-approval flexibility. In ANDA filings, QbD-based development is particularly relevant for demonstrating equivalence, robustness, and consistent performance of generic products. Similarly, NDA submissions increasingly reflect lifecycle-oriented QbD approaches, supporting post-approval change management and continuous improvement. As a result, QbD is transitioning from a recommended development philosophy to an implicit regulatory expectation in contemporary pharmaceutical submissions.
Recent regulatory guidance emphasizes the incorporation of Established Conditions (ECs) and Post-Approval Change Management Protocols (PACMPs) as part of a science- and risk-based QbD approach. These concepts, formalized under ICH Q12 (Pharmaceutical Product Lifecycle Management), enable manufacturers to manage post-approval changes efficiently within an approved design space. Regulatory agencies increasingly expect ANDA and NDA submissions to demonstrate clear linkage between QTPP, CQAs, CPPs, and control strategies, thereby supporting enhanced regulatory confidence and lifecycle flexibility.
Objectives
Primary Objectives
To critically evaluate peer-reviewed research studies, regulatory guidelines, and reported industrial practices that apply Quality by Design principles in the development of pharmaceutical formulations, encompassing both small-molecule and biopharmaceutical products.
To identify and assess the application of key QbD elements, including Quality Target Product Profile (QTPP), Critical Quality Attributes (CQAs), Critical Material Attributes (CMAs), Critical Process Parameters (CPPs), risk assessment techniques, Design of Experiments (DoE), and Process Analytical Technology (PAT), across different stages of formulation and process development.
To examine the influence of QbD adoption on product quality, manufacturing efficiency, regulatory flexibility, and lifecycle management, while identifying challenges and barriers that limit broader industrial implementation.
Key Principles of QbD
Design Space
Definition, significance, and flexibility
Design space is defined as the multidimensional combination of input variables (material attributes and process parameters) that have been demonstrated to assure product quality. Operating within the approved design space provides regulatory flexibility and does not require post-approval regulatory submissions.
Establishing & characterizing design space
Design space is established using scientific understanding, DoE, multivariate analysis, and risk assessment. It links formulation variables and process parameters directly to CQAs, ensuring robust product performance.
Adaptability in manufacturing
Design space enables manufacturers to make process adjustments within predefined limits, improving scalability, robustness, and adaptability without compromising quality.
Critical Material Attributes (CMAs)
Critical Material Attributes are the physical, chemical, biological, or microbiological properties of input materials that can significantly impact product quality. In pharmaceutical formulation development, CMAs commonly include excipient grade, particle size distribution, polymorphic form, moisture content, and supplier variability. Identification and control of CMAs are essential for ensuring formulation robustness, particularly in the context of raw material variability and global sourcing. Within the QbD framework, systematic evaluation of CMAs through risk assessment and experimental studies supports consistent product performance and strengthens supplier qualification strategies.
Control Strategy in QbD Framework
A control strategy is a planned set of controls, derived from current product and process understanding, that ensures consistent product quality throughout the lifecycle. According to ICH Q8(R2) and ICH Q10, the control strategy integrates material attributes, process parameters, in-process controls, finished product specifications, and real-time monitoring tools to maintain Critical Quality Attributes within predefined limits.
The control strategy is established based on systematic identification of Critical Material Attributes and Critical Process Parameters during formulation and process development. Material controls include specifications for raw materials and excipients, while process controls encompass operational parameters such as mixing time, granulation endpoint, compression force, and coating conditions. In-process testing and Process Analytical Technology tools are increasingly incorporated to enable continuous monitoring and early detection of deviations.
A robust control strategy is closely linked to the established design space. When operations are conducted within the approved design space, the control strategy ensures predictable performance without the need for additional regulatory submissions. This integrated approach supports regulatory flexibility, enhances process robustness, and enables effective lifecycle management through continual improvement and post-approval change management.
Knowledge Management and Lifecycle Learning
Knowledge management is a core expectation under ICH Q10 and plays a critical role in effective Quality by Design implementation. It involves systematic capture, analysis, storage, and reuse of product and process knowledge generated during pharmaceutical development and manufacturing. Development reports, risk assessments, Design of Experiments data, and control strategy justifications collectively form a structured knowledge base that supports informed decision-making.
Effective knowledge management enables continual learning by allowing manufacturers to leverage historical data for process optimization, deviation investigation, and lifecycle improvements. Data reuse across development, scale-up, and commercial manufacturing enhances consistency and reduces redundancy in experimental efforts. Within a QbD framework, knowledge management ensures that scientific understanding evolves throughout the product lifecycle, supporting regulatory compliance, post-approval change management, and continuous improvement initiatives.
Risk Assessment
CQAs are physical, chemical, biological, or microbiological properties that must be controlled to ensure product quality. CPPs are process parameters that have a direct and significant impact on CQAs.
Common tools include Failure Mode and Effects Analysis (FMEA), Ishikawa (fishbone) diagrams, Hazard Analysis and Critical Control Points (HACCP), and risk ranking and filtering. These tools help prioritize variables and guide experimental design.
Risk-based thinking shifts quality assurance from reactive testing to proactive design, minimizing failures and ensuring consistent product performance.
Risk assessment within a QbD framework is not a static activity but a dynamic and iterative process that extends throughout the product lifecycle. Risks identified during development are reassessed during scale-up, technology transfer, and commercial manufacturing based on accumulated process knowledge. Integration of risk management with Corrective and Preventive Action (CAPA) systems and knowledge management ensures continual improvement and aligns with the expectations of ICH Q10 Pharmaceutical Quality System.
Real-Time Release Testing (RTRT)
Implementation and integration in QbD
RTRT uses in-process data and PAT tools to ensure product quality, reducing reliance on traditional end-product testing.

Fig.2. QbD implementation framework
Real-time release testing (RTRT) represents a key operational component of the Quality by Design framework by enabling real-time, data-driven quality assurance during pharmaceutical manufacturing. RTRT relies on in-process measurements obtained through Process Analytical Technology tools rather than exclusive dependence on traditional end-product testing. This approach significantly reduces batch release timelines while allowing early identification of deviations, thereby minimizing the risk of batch failure.
Moreover, RTRT enhances overall process understanding and supports continuous monitoring of critical quality attributes throughout manufacturing. By embedding quality verification directly into the process, RTRT ensures batch-to-batch consistency and aligns closely with QbD objectives of proactive quality control, operational efficiency, and regulatory flexibility.
Benefits
Role in ensuring quality and consistency
RTRT supports continuous monitoring and control, ensuring batch-to-batch consistency and alignment with QbD objectives.
Outcomes of QbD Implementation
Implementation of Quality by Design in pharmaceutical formulation development consistently leads to measurable improvements in product quality, manufacturing robustness, and process predictability. Through systematic identification and control of critical material attributes and process parameters, QbD-based development minimizes formulation and process variability and enhances batch-to-batch consistency.
In addition, QbD facilitates efficient scale-up, technology transfer, and lifecycle management by establishing scientifically justified operating ranges and risk-based control strategies. These outcomes collectively support regulatory compliance, reduce manufacturing failures, and promote sustainable and efficient pharmaceutical production across the product lifecycle.
Quantitative outcomes reported across studies indicated a 30–50% reduction in experimental trials compared to conventional development approaches. Several studies documented 20–40% reductions in batch failures and rework, along with improved first-pass yield during scale-up. QbD-based development also supported faster and more predictable scale-up timelines, reducing delays during technology transfer and commercial manufacturing.
Challenges and Opportunities
Despite its demonstrated advantages, the implementation of Quality by Design presents several challenges, particularly during early stages of pharmaceutical development. QbD requires substantial initial investment in terms of time, experimental effort, and financial resources. The complexity of multivariate experimental designs and advanced statistical analyses necessitates specialized expertise, which may be limited in small- and medium-scale pharmaceutical organizations. Additionally, resistance to organizational change and inadequate training infrastructure can further impede widespread adoption.
Conversely, QbD offers significant long-term opportunities for the pharmaceutical industry. Continuous improvement through iterative process optimization enhances manufacturing robustness and reduces long-term operational costs. Improved process understanding supports faster development timelines, encourages innovation, and strengthens regulatory confidence. Furthermore, increasing global regulatory alignment and harmonization favor QbD-based submissions, positioning the approach as a strategic enabler for efficient international product development and approval.
Regulatory Acceptance and Practical Implementation of RTRT
Regulatory agencies, particularly the US Food and Drug Administration, actively support Real-Time Release Testing as part of a science- and risk-based Quality by Design approach. The FDA’s Process Analytical Technology initiative recognizes RTRT as an acceptable alternative to conventional end-product testing for solid oral dosage forms when supported by adequate process understanding and validated analytical models.
Across the included studies, RTRT was most frequently applied in solid oral dosage form manufacturing, where critical attributes such as blend uniformity, tablet hardness, and dissolution performance were monitored using in-line or on-line PAT tools. Commonly reported analytical techniques included near-infrared (NIR) spectroscopy, Raman spectroscopy, particle size analyzers, and multivariate statistical models for real-time data interpretation.
Despite its advantages, validation of RTRT remains a significant challenge. Key limitations include model robustness, calibration transfer across equipment, handling of process variability, and regulatory expectations for lifecycle model maintenance. Nevertheless, studies consistently demonstrated that when properly implemented, RTRT reduces batch release timelines, minimizes product rejections, and enhances batch-to-batch consistency.
Limitations of Real-Time Release Testing
Despite its advantages, implementation of Real-Time Release Testing presents several challenges. Model drift over time, variability in raw materials, and changes in process conditions may affect the robustness of chemometric models. Continuous model maintenance, calibration transfer, and periodic revalidation are therefore required. In addition, regulatory acceptance of RTRT depends heavily on data integrity and compliance with ALCOA+ principles, necessitating robust data governance and lifecycle management of analytical models.
Applications of QbD
Case Studies
QbD has been applied to optimize excipient levels, compression force, and granulation parameters, leading to improved dissolution, content uniformity, and mechanical strength.
In biologics, QbD aids in controlling critical parameters such as cell culture conditions, purification steps, and protein stability, improving yield and product consistency.
QbD-based approaches have reduced formulation variability and enhanced scale-up success in tablets and capsules.
QbD has also been successfully applied to topical and transdermal drug delivery systems, where control of rheological properties, drug release, and skin permeation is critical. In inhalation products, QbD facilitates optimization of particle size distribution, aerosol performance, and device compatibility. Furthermore, application of QbD in fixed-dose combinations (FDCs) supports systematic evaluation of drug–drug and drug–excipient interactions, ensuring consistent performance and regulatory compliance.
Table1. Application of QbD Across Pharmaceutical Dosage Forms
|
Dosage Form |
Typical CQAs |
Key CPPs |
QbD Tools Used |
|
Immediate-release tablets |
Dissolution, content uniformity, hardness |
Compression force, blending time |
QTPP, DoE, FMEA |
|
Modified-release tablets |
Drug release profile, friability |
Polymer concentration, coating parameters |
DoE, design space, risk assessment |
|
Capsules |
Content uniformity, disintegration |
Filling speed, lubricant level |
QTPP, DoE |
|
Nanoformulations |
Particle size, zeta potential, encapsulation efficiency |
Homogenization pressure, surfactant ratio |
DoE, CMA–CPP mapping |
|
Parenteral formulations |
Sterility, particle size, assay |
Filtration parameters, filling conditions |
Risk assessment, control strategy |
|
Biopharmaceuticals |
Potency, purity, stability |
Cell culture conditions, purification steps |
QbD framework, risk management |
Outcomes
Regulatory Landscape
- ICH guidelines
Recent regulatory developments further strengthen analytical aspects of QbD through ICH Q14 (Analytical Procedure Development) and ICH Q2(R2) (Validation of Analytical Procedures). These guidelines emphasize systematic method development, risk-based validation, and lifecycle management of analytical procedures, aligning analytical QbD with overall pharmaceutical development strategies.
- FDA quality systems approach
The FDA promotes QbD to enhance product understanding, reduce regulatory burden, and support science-based decision-making.
- Integration of QbD in regulatory submissions
Regulatory filings increasingly include QTPP, identified CQAs, design space justification, and risk-based control strategies.
- Global harmonization and convergence
QbD facilitates consistent regulatory expectations across regions, supporting international product approvals.
REGIONAL PERSPECTIVES
United States Food and Drug Administration (US FDA)
The US FDA promotes Quality by Design through its Pharmaceutical Quality for the 21st Century initiative and Process Analytical Technology framework. QbD principles are increasingly reflected in New Drug Applications and Abbreviated New Drug Applications, where applicants are encouraged to define QTPP, identify CQAs, justify design space, and implement risk-based control strategies. FDA guidance documents emphasize that robust process understanding can reduce regulatory burden and facilitate post-approval changes.
European Medicines Agency (EMA)
The EMA supports QbD implementation through scientific advice procedures and alignment with ICH guidelines. Regulatory submissions incorporating design space and enhanced process understanding are viewed favorably, particularly for complex and modified-release dosage forms. EMA encourages lifecycle management approaches that integrate QbD principles to ensure sustained product quality.
World Health Organization (WHO)
The WHO recognizes QbD as a valuable tool for improving pharmaceutical quality, particularly in low- and middle-income countries. Adoption of QbD principles supports consistent manufacturing, reduces variability, and strengthens regulatory oversight. WHO guidelines increasingly emphasize risk-based approaches aligned with ICH standards.
Indian Regulatory Perspective (CDSCO)
Although explicit QbD mandates are limited, the Central Drugs Standard Control Organization increasingly aligns with ICH guidelines. Indian manufacturers adopting QbD principles demonstrate improved readiness for global regulatory submissions, especially for export-oriented products. QbD-based development supports compliance with evolving regulatory expectations and international harmonization.
GLOBAL CONTEXT
India remains a major global supplier of generic medicines, with a significant proportion of pharmaceutical exports directed toward regulated markets such as the United States and Europe. Consequently, Indian manufacturers increasingly adopt QbD principles to meet USFDA ANDA requirements and inspection expectations. Emphasis on quality culture, data integrity, and science-based development has become critical for maintaining regulatory compliance and sustaining international market access.
CHALLENGES AND OPPORTUNITIES
Challenges
Opportunities
Continuous Improvement
- Iterative process optimization and feedback loops
QbD supports lifecycle management through continual monitoring, data evaluation, and refinement of processes.
- Risk mitigation and CAPA
Ongoing risk assessments and Corrective and Preventive Actions (CAPA) enable proactive problem resolution.
- Data-driven decision-making and training
Effective QbD implementation relies on data analytics, knowledge management, and continuous workforce training.
FUTURE PERSPECTIVES
Integration of PAT, spectroscopy, and omics technologies enables deeper process understanding and real-time quality control.
Artificial intelligence and machine learning facilitate predictive process control, optimization, and faster decision-making.

Fig.3. Integration of PAT, AI, and continuous manufacturing with QbD
Use of IoT, Big Data, and cyber-physical systems enables automated, connected, and intelligent pharmaceutical manufacturing.
End-to-end visibility improves quality assurance, traceability, and supply chain resilience.
Global Harmonization
- ICH-led initiatives
Standardized terminology, shared regulatory expectations, and collaborative initiatives promote consistent global implementation of QbD.
- Joint regulatory inspections
Increased collaboration among regulatory agencies supports efficient oversight and mutual recognition.
MATERIALS AND METHODS
Review Protocol
This systematic review was conducted in accordance with the Preferred Reporting Items for Systematic Reviews and Meta-Analyses (PRISMA) guidelines. A predefined review protocol was established prior to the initiation of the study, outlining the objectives, search strategy, eligibility criteria, study selection process, and data extraction methodology. The protocol was designed to ensure transparency, reproducibility, and methodological rigor.
Search Strategy
A comprehensive and systematic literature search was performed across multiple electronic databases, including PubMed, Google Scholar, ScienceDirect, SpringerLink, and Wiley Online Library. The search strategy employed a combination of controlled vocabulary and free-text terms related to Quality by Design and pharmaceutical formulation development. Boolean operators were used to refine the search.
The primary keywords included:
An example search string used was:“Quality by Design” OR QbD) AND (“pharmaceutical formulation” OR “formulation development”
Eligibility Criteria
Inclusion Criteria
Exclusion Criteria
Study Selection
All retrieved records were imported into reference management software, and duplicate entries were removed. Initial screening was performed based on titles and abstracts to exclude irrelevant studies. Full-text articles of potentially eligible studies were then assessed against the inclusion and exclusion criteria. Only studies meeting all eligibility criteria were included in the final qualitative synthesis.
Data Extraction and Synthesis
Data were independently extracted from the included studies using a predefined data extraction form. The extracted information included:
Due to heterogeneity in study designs and outcomes, data were synthesized qualitatively and organized into descriptive tables and thematic categories.
PRISMA DATA
Study Selection Results
The literature search identified 642 records across all databases. After removal of duplicates (n=146), 496 records were screened based on titles and abstracts. Of these, 546 records were excluded due to irrelevance. Full-text assessment was conducted for 96 articles, resulting in 58 studies being included in the final qualitative synthesis.
Risk of Bias and Quality Assessment
Formal quantitative risk-of-bias assessment was not performed due to heterogeneity in study designs and outcome measures. However, methodological rigor was qualitatively assessed based on clarity of QbD implementation, appropriateness of experimental design, and alignment with regulatory guidance.
RESULTS
Study Selection
The systematic database search yielded a substantial number of records across the selected databases. Following removal of duplicates and screening of titles and abstracts, a subset of articles was subjected to full-text evaluation. Based on predefined eligibility criteria, a final group of studies was included in the qualitative synthesis. The study selection process is summarized using a PRISMA flow diagram.
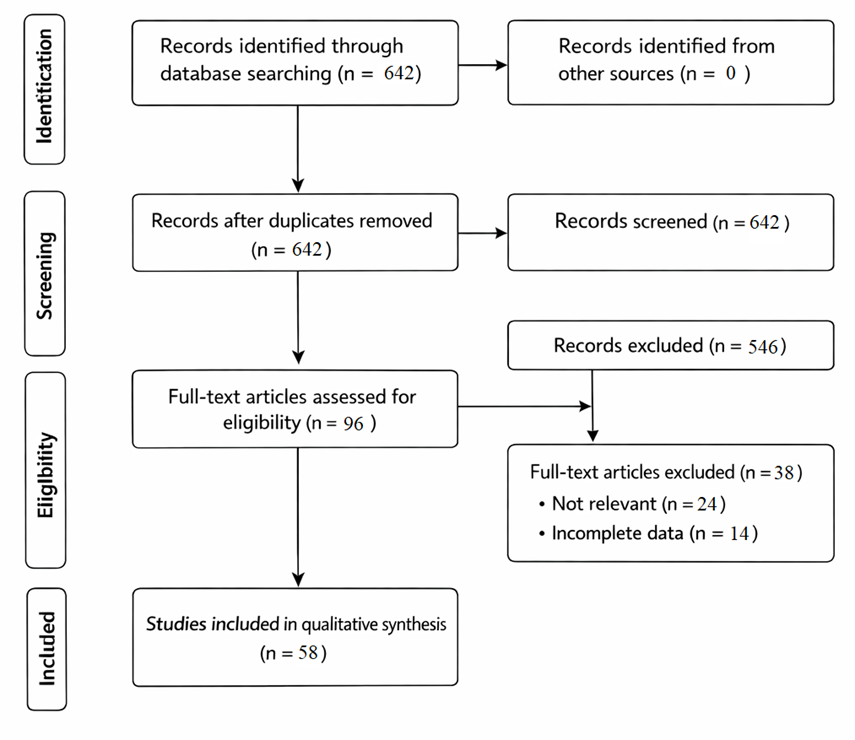
Figure 4. PRISMA flow diagram illustrating the study selection process.
QUANTITATIVE MAPPING (DATA STATEMENT)
Across the included studies, approximately 70–80% employed Design of Experiments as a primary QbD tool. Definition of design space was reported in 45–60% of studies, primarily in modified-release and complex dosage forms. Use of Process Analytical Technology and Real-Time Release Testing was observed in 20–30% of studies, reflecting gradual adoption of advanced analytical tools in formulation development.
Characteristics of Included Studies
The included studies were published between 2010 and 2025 and represented a wide range of pharmaceutical formulation research. The majority of studies involved experimental formulation development, with additional case studies reporting pilot-scale or industrial-scale applications.
Dosage forms investigated included:
Studies were conducted at laboratory, pilot, and industrial scales, highlighting the applicability of QbD across different stages of product development.
QbD Tools and Methodologies Applied
Across the included studies, a consistent framework of QbD tools was observed:
DoE emerged as the most frequently applied tool, reflecting its effectiveness in reducing experimental trials and improving process understanding.
Application Across Dosage Forms
QbD approaches enabled optimization of granulation parameters, compression force, and coating conditions, resulting in improved dissolution behavior, content uniformity, and batch consistency.
Studies demonstrated that QbD facilitated the achievement of target release profiles through systematic optimization of polymer type, matrix composition, and processing conditions.
Application of QbD resulted in controlled particle size, improved encapsulation efficiency, and enhanced reproducibility, addressing common challenges associated with nanoscale delivery systems.
QbD-based development focused on sterility assurance, particle size control, and critical processing steps, contributing to improved safety and product consistency.
Key Outcomes of QbD Implementation
The collective findings of the included studies demonstrated that QbD-based formulation development led to:
Regulatory Relevance
Most studies aligned with ICH Q8, Q9, and Q10 guidelines, with several reporting the successful submission of design space and control strategies as part of regulatory filings. QbD-based documentation facilitated Emerging regulatory interest, particularly for complex and modified-release dosage forms, and supported effective lifecycle management.
Table 2. Frequency of QbD Tool Usage Across Included Studies
|
QbD Tool |
Approximate Usage (%) |
|
Design of Experiments (DoE) |
70–80% |
|
Risk Assessment (FMEA, Ishikawa) |
65–75% |
|
Design Space Establishment |
45–60% |
|
Process Analytical Technology (PAT) |
25–30% |
|
Real-Time Release Testing (RTRT) |
20–25% |
Table 3: Summary of Studies Included in the Systematic Review
|
Author (Year) |
Dosage Form |
QbD Tools Applied |
Key CQAs |
Major Outcomes |
Regulatory Relevance |
|
Yu et al. (2008) |
Solid oral |
QTPP, DoE, Risk assessment |
Dissolution, assay, content uniformity |
Improved process understanding and robustness |
ICH Q8-compliant |
|
Beg et al. (2019) |
Modified-release tablets |
QTPP, FMEA, DoE |
Drug release, hardness |
Reduced variability, optimized formulation |
Supports design space submission |
|
Mishra et al. (2020) |
Nanoparticles |
CQAs, CMAs, DoE |
Particle size, zeta potential |
Enhanced product quality and stability |
Facilitates regulatory flexibility |
|
Patel et al. (2021) |
Immediate-release tablets |
Risk assessment, DoE |
Disintegration, friability |
Robust formulation with fewer batch failures |
Lifecycle management |
|
Singh et al. (2022) |
Sustained-release system |
QTPP, CPPs, PAT |
Drug release kinetics |
Improved control strategy |
Aligns with ICH Q10 |
|
Rao et al. (2023) |
Parenteral formulation |
Risk analysis, DoE |
Sterility, particle size |
Enhanced safety and consistency |
Emerging regulatory interest improvement |
Table 4: Common QbD Tools and Their Purpose
|
QbD Tool |
Purpose |
|
QTPP |
Defines target product quality characteristics |
|
CQAs |
Identifies attributes affecting product quality |
|
Risk Assessment |
Prioritizes CMAs and CPPs |
|
DoE |
Establishes relationship between variables |
|
Design Space |
Ensures operational flexibility |
|
Control Strategy |
Maintains consistent product quality |
Table 5: Advantages of QbD Compared to Conventional Development
|
Aspect |
Conventional |
QbD |
|
Process understanding |
Limited |
High |
|
Regulatory flexibility |
Low |
High |
|
Risk management |
Reactive |
Proactive |
|
Scale-up success |
Variable |
Predictable |
Table 6. QbD Tools and Their Regulatory Purpose
|
QbD Tool |
Regulatory Purpose |
|
QTPP |
Defines product intent |
|
CQAs |
Identifies quality risks |
|
Risk assessment |
Justifies experimental focus |
|
Design space |
Enables regulatory flexibility |
|
Control strategy |
Ensures consistent quality |
|
RTRT |
Supports real-time compliance |
Table 7. Challenges in QbD Implementation and Mitigation Strategies
|
Challenge |
Mitigation |
|
High initial cost |
Lifecycle cost-benefit |
|
Statistical complexity |
Training & software |
|
Regulatory uncertainty |
Early scientific advice |
|
Data overload |
Knowledge management systems |
DISCUSSION
Impact of QbD on Formulation Quality and Process Understanding
The findings of this systematic review demonstrate that the application of Quality by Design significantly enhances formulation robustness and process understanding across diverse pharmaceutical dosage forms. By systematically identifying and controlling critical formulation and process variables, QbD enables consistent achievement of predefined quality attributes, thereby reducing variability and improving product performance. The reviewed studies consistently reported improved reproducibility, reduced batch failures, and enhanced predictability during scale-up and technology transfer.
Unlike conventional empirical approaches, QbD establishes a clear scientific rationale linking material attributes and process parameters to critical quality attributes. This comprehensive understanding supports proactive quality assurance and minimizes the likelihood of unexpected deviations during commercial manufacturing.
Academia versus Industry Adoption of QbD
The review indicates that academic research predominantly focuses on demonstrating the applicability of individual QbD tools, particularly DoE and risk assessment, at laboratory scale. In contrast, industrial implementation emphasizes lifecycle management, regulatory compliance, and scalability. Despite regulatory encouragement, industrial adoption remains slower due to higher resource requirements, organizational inertia, and the need for cross-functional expertise. Bridging this gap requires stronger collaboration between academia, industry, and regulatory bodies.
Scientific Risk versus Regulatory Risk
Scientific risk arises from inadequate understanding or control of Critical Quality Attributes, leading to product variability or failure. Regulatory risk, however, stems from insufficient scientific justification, incomplete documentation, or inability to demonstrate control over CPPs and design space. QbD mitigates both risks by embedding scientific rationale into regulatory submissions, thereby strengthening regulatory confidence and reducing post-approval uncertainties.
Role of Risk Assessment and Design of Experiments
Risk assessment tools such as Failure Mode and Effects Analysis and Ishikawa diagrams were widely employed to identify Critical Material Attributes and Critical Process Parameters during early development stages. These tools enabled prioritization of high-risk variables, allowing efficient allocation of experimental resources. Design of Experiments emerged as a cornerstone of QbD implementation, facilitating simultaneous evaluation of multiple variables and their interactions.
Compared to traditional one-factor-at-a-time experimentation, DoE-based approaches reduced the number of experimental runs while providing statistically meaningful insights into formulation and process behavior. This multivariate understanding was essential for defining design space and developing robust control strategies.
Comparison with Conventional Development Approaches
Conventional pharmaceutical development approaches often rely on extensive end-product testing and conservative operating limits, resulting in limited flexibility and inefficient manufacturing practices. In contrast, QbD-driven development integrates quality considerations into product and process design, enabling the establishment of scientifically justified design space. Operating within an approved design space allows manufacturers to make process adjustments without additional regulatory submissions, thereby enhancing operational flexibility and manufacturing efficiency.
This shift from reactive quality control to proactive quality design represents a fundamental transformation in pharmaceutical development philosophy.
Regulatory and Industry Perspective
The integration of QbD principles into regulatory submissions has been shown to improve regulatory confidence and facilitate approval processes. Alignment with ICH guidelines Q8, Q9, Q10, and Q11 provides a harmonized framework that supports lifecycle management and post-approval change management. Regulatory agencies increasingly recognize the value of QbD in ensuring consistent product quality, particularly for complex dosage forms and advanced drug delivery systems.
Despite regulatory encouragement, industrial adoption of QbD remains uneven. Small- and medium-scale manufacturers often face challenges related to limited technical expertise, high initial investment costs, and insufficient access to advanced analytical technologies such as Process Analytical Technology and real-time release testing.
Challenges and Limitations
Although QbD offers significant advantages, its implementation is associated with several challenges. These include the need for specialized statistical expertise, increased data generation and management requirements, and substantial upfront investment in experimental design and analytical tools. Additionally, limited integration of PAT and digital technologies restricts the full realization of real-time quality assurance in many development settings.
From a regulatory perspective, uncertainty regarding the acceptance of emerging tools such as artificial intelligence-based models and advanced predictive analytics further limits widespread implementation, underscoring the need for clearer regulatory guidance and harmonized evaluation frameworks.
Research Gaps and Future Directions
This review identified several research gaps that warrant further investigation. Limited application of QbD in biologics, biosimilars, personalized medicines, and highly complex dosage forms highlights the need for expanded research in these areas. Furthermore, integration of QbD with artificial intelligence, machine learning, and digital Process Analytical Technology remains underexplored.
Future research should focus on expanding QbD adoption in continuous manufacturing, developing standardized approaches for digital model validation, and enhancing capacity building to support broader industrial implementation.
Significant research gaps persist in the application of QbD to biosimilars, gene and RNA-based therapies, personalized medicines, and continuous manufacturing systems. These emerging areas involve higher complexity, increased variability, and evolving regulatory expectations. Furthermore, validation strategies for digital models, PAT tools, and continuous processes remain insufficiently standardized, highlighting the need for further research and regulatory guidance.
CONCLUSION
Quality by Design represents a robust, science-based, and regulatory-aligned framework that significantly improves pharmaceutical formulation development by enhancing product quality, process understanding, and manufacturing robustness. This systematic review demonstrates that QbD-based approaches reduce variability, support efficient scale-up, and enable risk-based regulatory flexibility across a wide range of dosage forms.
Despite challenges related to resource requirements and technical expertise, the long-term benefits of QbD—including improved lifecycle management, reduced manufacturing risk, and enhanced regulatory confidence—outweigh its initial implementation barriers. As pharmaceutical products continue to increase in complexity, the integration of QbD with advanced analytical technologies, artificial intelligence, and continuous manufacturing is expected to play a pivotal role in shaping the future of pharmaceutical development. Wider adoption of QbD will be essential for achieving efficient, predictable, and patient-centric drug development aligned with global regulatory expectations.
With increasing product complexity and regulatory expectations, Quality by Design is transitioning from a recommended approach to a practical necessity in pharmaceutical development. Future integration of QbD with digital twins, artificial intelligence, and data-driven design space optimization is expected to further enhance predictability, efficiency, and patient-centric product development.
REFERENCES



Prajwal Gosavi, Akanksha Nirmal, Rutuja Ahire, Quality By Design (QbD) in Pharmaceutical Formulation Development: A Systematic Review, Int. J. of Pharm. Sci., 2026, Vol 4, Issue 1, 528-548. https://doi.org/10.5281/zenodo.18165771
 10.5281/zenodo.18165771
10.5281/zenodo.18165771