
We use cookies to ensure our website works properly and to personalise your experience. Cookies policy
Department of Pharmaceutical Analysis & Chemistry, Qis College of Pharmacy, Ongole.
Quality by Design (QbD) has emerged as a transformative approach in pharmaceutical development, aiming to build quality into products from the earliest stages. Unlike traditional quality assurance methods that rely heavily on end-product testing, QbD emphasizes a thorough understanding of processes, identification of critical quality attributes (CQAs), and control strategies based on risk assessment and scientific rationale. This review explores the fundamental principles of QbD, its application across various stages of pharmaceutical development, and the regulatory framework supporting its implementation. Key components such as Design of Experiments (DoE), risk management tools, and the use of process analytical technology (PAT) are discussed in detail. The article also highlights the benefits and challenges of adopting QbD, including improved product consistency, regulatory flexibility, and resource optimization. Through this comprehensive overview, the review aims to provide insights into the evolving landscape of pharmaceutical quality management and the pivotal role of QbD in ensuring patient safety and product efficiency. Definition: Quality by Design (QbD) is a systematic approach to pharmaceutical development that emphasizes designing and building quality into a product from the beginning and process understanding and controls based on sound science Quality risk management (ICHQ8)
Quality by Design (QbD) is a systematic, science-based approach to pharmaceutical development that emphasizes building quality into a product from the earliest stages, rather than relying solely on end-product testing. The concept was introduced by the U.S. Food and Drug Administration (FDA) as part of its initiative to modernize pharmaceutical manufacturing and ensure consistent product performance. QbD aims to understand how formulation components, process parameters, and material attributes collectively influence the critical quality attributes (CQAs) of a drug product. The foundation of QbD lies in the principle that quality should be designed into a product rather than tested into it. This approach integrates comprehensive risk assessment, experimental design, and process understanding to ensure that the final product consistently meets its intended performance. Key elements of QbD include defining a Quality Target Product Profile (QTPP), identifying Critical Quality Attributes (CQAs), determining Critical Material Attributes (CMAs) and Critical Process Parameters (CPPs), and establishing a design space within which consistent quality can be achieved. By applying QbD principles, pharmaceutical companies can develop more robust and efficient manufacturing processes. This not only enhances product quality and patient safety but also provides regulatory flexibility and facilitates continuous improvement. Regulatory agencies such as the FDA and the European Medicines Agency (EMA) encourage the adoption of QbD to promote innovation, reduce variability, and streamline product lifecycle management. In summary, QbD represents a paradigm shift from traditional quality assurance methods toward a proactive, knowledge-driven framework. It enables better control of product and process variability, ultimately leading to higher confidence in pharmaceutical quality and improved outcomes for patients.
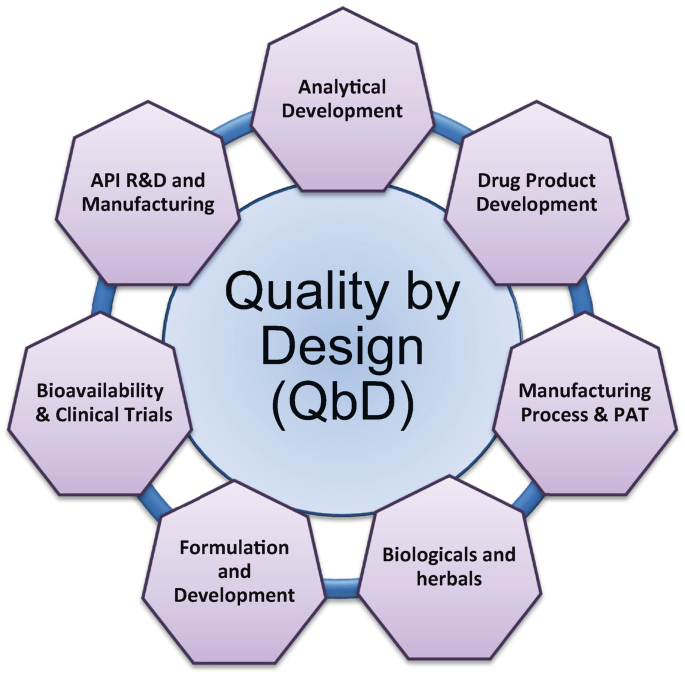
Concepts Of QBD:
Quality target product profile:
In the pharmaceutical development process, the Quality Target Product Profile (QTPP) serves as the cornerstone of the Quality by Design (QbD) approach. It outlines the critical quality attributes that define the desired performance, safety, and efficacy of a pharmaceutical product. QTPP provides a prospective summary of the quality characteristics that the final product should possess to ensure it meets the intended clinical objectives and regulatory requirements.
Definition:
According to the U.S. Food and Drug Administration (FDA), the QTPP is “a prospective summary of the quality characteristics of a drug product that ideally will be achieved to ensure the desired quality, taking into account safety and efficacy.”
In simple terms, the QTPP acts as a blueprint for product development, guiding formulation scientists and process engineers in designing and controlling the manufacturing process.
Role of QTPP in QbD:
In the QbD framework, the QTPP is the starting point for systematic product and process development. It drives the identification of:
Critical Quality Attributes (CQAs) of the product,
Critical Material Attributes (CMAs) and Critical Process Parameters (CPPs), and
The overall control strategy to maintain consistent product quality.
By clearly defining the QTPP at the early development stage, teams can ensure that all subsequent steps align with the intended therapeutic purpose and patient needs.
Key Elements of a QTPP
A typical QTPP includes the following parameters, tailored to the dosage form and route of administration:
|
Parameter |
Description |
|
|
Dosage form |
Defines the physical form of the product (tablet, capsule, injection, etc.) |
|
|
Route of administration |
Specifies how the product will be delivered to the patient (oral, parenteral, topical, etc.) |
|
|
Dosage strength |
Indicates the amount of active pharmaceutical ingredient (API) per unit dose |
|
|
Intended use or indication |
Clarifies the therapeutic purpose of the product |
|
|
Bioavailability and pharmacokinetics |
Target levels to ensure desired therapeutic action |
|
|
Stability |
Desired shelf-life and storage conditions |
|
|
Impurity limits |
Acceptable levels of degradation products or residual solvents |
|
|
Appearance and organoleptic properties |
Includes color, taste, odor, and visual aspects |
|
|
Container closure system |
Must ensure product protection and compatibility |
|
|
Safety and efficacy profile |
Must be consistent with the intended therapeutic effect. |
|
Relationship Between QTPP and CQAs:
Each element of the QTPP translates into one or more Critical Quality Attributes (CQAs)—the physical, chemical, biological, or microbiological properties that must be controlled to ensure the product meets its QTPP.
For example:
Design Space:
Design Space (DS) is a fundamental concept in the Quality by Design (QbD) framework, as outlined by the International Council for Harmonisation (ICH) guideline Q8 (R2). It represents the multidimensional combination and interaction of input variables (e.g., material attributes) and process parameters that have been demonstrated to assure product quality. Operating within the established design space is not considered a change, while movement outside the design space requires regulatory post-approval notification.
Definition:
According to ICH Q8 (R2):
“Design Space is the multidimensional region that links material attributes and process parameters with product quality.” It is developed through systematic experimentation and data analysis, establishing a relationship between critical process parameters (CPPs) and critical quality attributes (CQAs).
Role in QBD
The purpose of establishing a design space is to achieve a thorough process understanding and enhance flexibility in manufacturing. It allows manufacturers to make adjustments within the defined region without additional regulatory approvals, provided the product quality remains within acceptable limits.
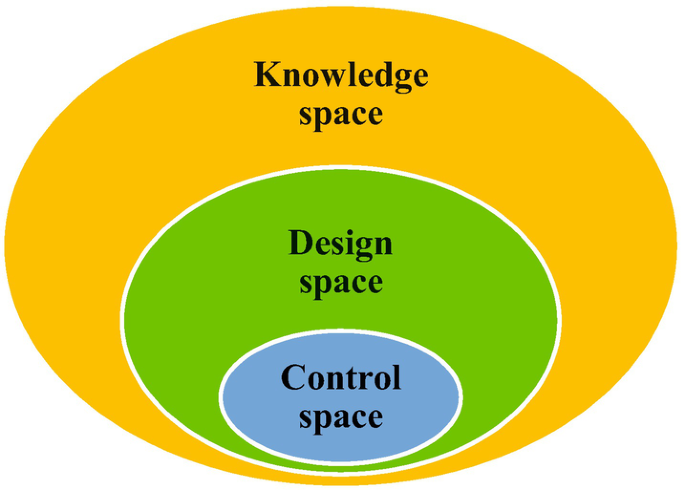
Control Strategy:
The control strategy is a fundamental component of the Quality by Design (QbD) framework in pharmaceutical development and manufacturing. It refers to a planned set of controls, derived from product and process understanding, that ensures the final product consistently meets its quality attributes and regulatory requirements throughout the lifecycle. According to the International Council for Harmonisation (ICH) guideline Q10 and Q11, a control strategy is “a planned set of controls, derived from current product and process understanding that ensures process performance and product quality.”
The primary objective of the control strategy is to maintain a state of control by managing variability in raw materials, process parameters, and environmental conditions, thereby assuring the quality of the drug product.
Product Life Cycle Management And Continual Improvement:
Product Life Cycle Management (PLM) in Quality by Design (QbD) :
Product Life Cycle Management (PLM) is a systematic approach that encompasses all stages of a pharmaceutical product’s existence — from conception, development, and manufacturing to marketing, post-approval monitoring, and eventual discontinuation. Within the Quality by Design (QbD) framework, PLM ensures that product quality is maintained consistently throughout its lifecycle, while also supporting regulatory compliance and efficient knowledge management.
Continual Improvement in Quality by Design (QbD) :
Continual improvement is a core principle of both QbD and the ICH Q10 Pharmaceutical Quality System. It refers to the ongoing effort to enhance product quality, process efficiency, and regulatory compliance through data-driven decision-making and scientific understanding.
Regulatory agencies encourage continual improvement within the QbD framework, as it demonstrates a proactive commitment to quality. The ICH guidelines (Q8–Q12) advocate a science- and risk-based approach that enables flexibility in post-approval changes when supported by strong process understanding.
The synergy between PLM and continual improvement forms the backbone of modern pharmaceutical quality systems. PLM provides the framework for structured data and knowledge flow across all lifecycle stages, while continual improvement drives the dynamic evolution of processes based on that knowledge. Together, they enable a closed-loop control system where product and process understanding are continually enhanced — resulting in sustained compliance, reduced risk, and consistent delivery of high-quality products.
Tools of QbD:
The concept of qbd has two components -the science underlying the design and science of manufacturing. Upon understanding the elements of QbD it is important to familiar with the commonly used tools in QbD are:
Risk Assessment:
Risk assessment is a fundamental element of the Quality by Design (QbD) framework, as outlined by the International Conference on Harmonisation (ICH) guidelines, particularly ICH Q8 (R2), Q9, and Q10. Within QbD, risk assessment serves as a systematic process for identifying, analysing, and controlling variables that could affect the quality, safety, and efficacy of pharmaceutical products. Its ultimate goal is to ensure that product quality is built into the design and development process rather than tested at the end.
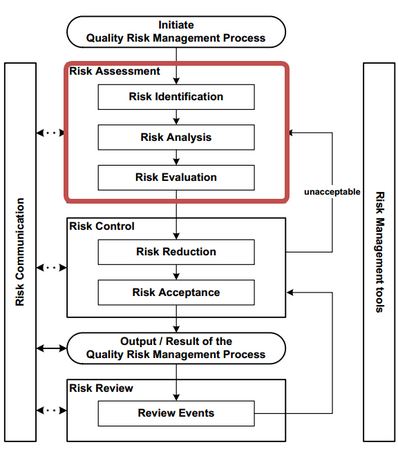
Steps in Risk Assessment Process
The risk assessment process in QbD typically includes three interrelated steps — risk identification, risk analysis, and risk evaluation — as recommended by ICH Q9.
Risk Identification
This step involves recognizing all possible variables, inputs, or process steps that may influence product quality. Sources of information may include prior knowledge, literature, laboratory studies, and manufacturing experience. Tools commonly used include:
Risk Analysis
In this step, each identified risk is analyzed to estimate the likelihood of occurrence, the severity of its impact, and its detectability. This quantitative or qualitative evaluation helps in prioritizing risks. For instance:
Risk Evaluation
Here, the significance of each risk is compared against predetermined criteria or thresholds to decide which risks require control or mitigation. The evaluation supports defining the control strategy — determining which parameters must be tightly controlled and which can be more flexible within the design space.
Tools and Techniques for Risk Assessment in QbD
Several structured and semi-quantitative tools are used for risk assessment:
|
Tool/Method |
Purpose |
Typical Application |
|
FMEA |
Identify and prioritize potential failure modes |
Manufacturing and formulation steps |
|
FTA |
Determine root causes of failures |
Complex system analysis |
|
Ishikawa Diagram |
Visualize cause–effect relationships |
Early-stage identification |
|
HAZOP (Hazard and Operability Study) |
Assess deviations from intended operation |
Process design and scale-up |
|
DOE (Design of Experiments) |
Quantify and validate impact of factors |
Establishing design space |
Integration of these tools helps maintain a scientific and transparent approach to risk management.
Design Of Experiment (DoE):
Design of Experiments (DoE) is a fundamental statistical and systematic tool used within the Quality by Design (QbD) framework to understand the relationship between process variables and product quality attributes. It enables scientific product and process development by providing structured methods for exploring the influence of multiple factors simultaneously. Regulatory agencies such as the U.S. Food and Drug Administration (FDA) and the International Council for Harmonisation (ICH) recognize DoE as a critical element in establishing a robust design space, as outlined in ICH Q8 (R2).
Principles of DoE:
The foundation of DoE lies in three core principles:
These principles enable reliable statistical inference and improve the robustness of conclusions drawn from the experiments.
Data Analysis and Model Validation:
Statistical software (e.g., Design-Expert®, JMP®, Minitab®) is often used for data fitting and model validation. Model adequacy is verified through:
Validated models provide a quantitative understanding of the process and support risk-based decision-making.
Regulatory Perspective:
Regulatory guidelines such as ICH Q8 (R2), ICH Q9, and ICH Q10 encourage the use of DoE for pharmaceutical development. FDA’s Process Analytical Technology (PAT) framework also aligns with DoE-based approaches for real-time process control. Submitting DoE results in regulatory filings demonstrates scientific process understanding and facilitates flexible post-approval changes within the established design space.
Process Analytical Technology (PAT):
Pharmaceutical manufacturing has evolved from traditional end-product testing toward a science- and risk-based approach emphasizing product and process understanding. The Quality by Design (QbD) framework, introduced by the International Council for Harmonisation (ICH Q8–Q11), promotes the design of quality into products from the earliest stages of development. Within this framework, Process Analytical Technology (PAT) plays a crucial role as an enabling tool for achieving real-time process monitoring, control, and continuous improvement.
Concept of Process Analytical Technology (PAT) :
PAT is defined by the U.S. Food and Drug Administration (FDA) as “a system for designing, analysing, and controlling manufacturing through timely measurements of critical quality and performance attributes of raw and in-process materials and processes, to ensure final product quality.” PAT combines analytical tools, process analysers, data acquisition systems, and multivariate data analysis to monitor key process parameters (CPPs) and critical quality attributes (CQAs) in real time. The goal is to move from empirical batch testing toward real-time process control and assurance of quality.
Common PAT Tools and Techniques:
|
Technique |
Measurement Type |
Application |
|
NIR Spectroscopy |
Chemical composition |
Blend uniformity, drying processes |
|
Raman Spectroscopy |
Molecular structure |
Crystallinity, polymorphism |
|
FTIR Spectroscopy |
Functional group analysis |
Reaction monitoring |
|
UV–Vis Spectroscopy |
Concentration analysis |
Dissolution, content uniformity |
|
Mass Spectrometry (MS) |
Molecular weight, impurities |
Biopharmaceutical process monitoring |
|
Particle Size Analyzers (Laser Diffraction) |
Physical attributes |
Granulation, milling |
Challenges:
Challenges Facing Quality by Design (QbD)
APPLICATIONS OF QBD:
Quality by Design (QbD) is a systematic approach used in the pharmaceutical and biotechnology industries to ensure the quality, safety, and effectiveness of products. It focuses on understanding processes and controlling variables from the beginning of development. Some important applications include:
QbD helps design robust formulations by identifying critical material attributes and process parameters that affect product quality.
It allows efficient process development and optimization through design of experiments (DoE) and risk assessment tools.
QbD ensures reliable analytical methods by defining method performance characteristics and establishing control strategies.
QbD supports regulatory submissions by providing scientific justification and detailed process understanding required by agencies like the FDA and EMA.
By controlling variability, QbD minimizes production failures, improves yield, and reduces costs.
It encourages lifecycle management and continuous process verification, ensuring long-term product quality.
QbD is increasingly applied in biologics to ensure consistency in complex processes such as fermentation and purification.
CONCLUSION:
Quality by Design (QbD) represents a transformative approach to pharmaceutical development and manufacturing, shifting the focus from end-product testing to proactive quality assurance throughout the entire lifecycle of a product. By systematically integrating scientific understanding, risk assessment, and process control, QbD ensures that quality is built into the product from the very beginning rather than being tested into it at the end. This paradigm fosters a deep understanding of the relationships between critical quality attributes (CQAs), critical process parameters (CPPs), and material attributes, thereby promoting robust and consistent product performance. The implementation of QbD enhances regulatory flexibility, improves process efficiency, and reduces overall production costs by minimizing batch failures and process variability. Furthermore, it aligns with the regulatory expectations set by ICH guidelines (Q8–Q11), which emphasize a science- and risk-based framework for pharmaceutical quality. The use of advanced analytical tools, design of experiments (DoE), and process analytical technology (PAT) enables real-time monitoring and control, supporting continuous improvement and innovation.
REFERENCES


S. Deepthi*, Dr. M. Kishore babu, SK. Sheema, T. Sampath Kumar, P. Anjani Praneetha, R. Mashith, P. Lakshmitha, Quality by Design in Pharmaceutical Development: A Comprehensive Review of Concepts, Tools and Applications, Int. J. of Pharm. Sci., 2025, Vol 3, Issue 10, 2706-2718 https://doi.org/10.5281/zenodo.17442528
 10.5281/zenodo.17442528
10.5281/zenodo.17442528