
We use cookies to ensure our website works properly and to personalise your experience. Cookies policy
Pharma Quality Assurance Professional, Alumni, PES College of Pharmacy, 50 Feet Road, Hanumanthanagar, Banashankari Stage I, Bengaluru – 560050, Karnataka, India
Quality Management Systems (QMS) in the pharmaceutical industry establish the structured policies, procedures, and defined responsibilities required to maintain consistent product quality, safety, and efficacy throughout the entire product lifecycle. This review integrates the foundational regulatory expectations outlined in current Good Manufacturing Practices (cGMP), including FDA 21 CFR Parts 210, 211, and 11, alongside EU GMP Annex 11. It further incorporates the harmonized framework of ICH Q10, supported by Quality Risk Management and Knowledge Management, as well as recognized industry standards such as ISO 9001:2015 and ISPE GAMP 5. Key elements of an effective Pharmaceutical Quality System (PQS)—process performance and product quality monitoring, corrective and preventive actions (CAPA), change management, and management review—are examined in depth. Additional focus is placed on operationalizing QMS principles through robust data integrity practices, supplier and outsourced activity oversight, and the progressive transition from paper based or hybrid models to integrated electronic QMS (eQMS) platforms. The review also proposes a practical maturity pathway and identifies meaningful quality key performance indicators (KPIs) applicable to commercial manufacturing environments, supporting continual improvement and long term inspection readiness.
Pharmaceutical quality is both a legal mandate and an ethical responsibility intended to safeguard patient health by ensuring that every medicinal product consistently meets established standards of safety, efficacy, and performance. A robust Quality Management System (QMS) provides the structural foundation to achieve this objective by integrating organizational policies, defined processes, documented procedures, and the necessary resources to maintain a sustained state of control throughout the product lifecycle. Modern pharmaceutical quality practices are anchored in current Good Manufacturing Practice (cGMP) regulations, which establish fundamental expectations for manufacturing and quality oversight. These regulatory requirements are complemented by harmonized international guidance, most notably ICH Q10, which promotes a lifecycle?based pharmaceutical quality system focused on science?driven decision?making, continual improvement, and enhanced process understanding. Together, these frameworks enable organizations to strengthen compliance, reduce variability, support innovation, and ensure consistent delivery of high?quality medicinal products to patients.
MATERIALS & METHODS
This narrative review was developed through the structured evaluation of authoritative regulatory frameworks, international guidelines, and industry standards relevant to pharmaceutical Quality Management Systems (QMS). Key regulatory documents examined include FDA 21 CFR Parts 210, 211, and 11, as well as EU GMP Annex 11, which collectively define baseline requirements for manufacturing controls, quality operations, and computerized system compliance. Harmonized international guidelines—specifically ICH Q10, ICH Q9, and ICH Q8—were reviewed to provide additional context on lifecycle?based quality management, risk management principles, and pharmaceutical development strategies. Complementary standards such as ISO 9001:2015 and ISPE GAMP 5 were included to integrate broader quality management philosophies and validated approaches for computerized systems. Contemporary practitioner literature, including industry white papers and technical publications, was also assessed to align the review with current operational practices and technological advancements. As this work is based solely on published documents and secondary sources, no human or animal subjects were involved, and therefore no ethical approval was required.
RESULTS AND DISCUSSION
Objectives of a Pharmaceutical QMS
The primary objectives of a Pharmaceutical Quality Management System (QMS) are centered on ensuring that medicinal products consistently meet established standards of quality, safety, and efficacy. A well?implemented QMS seeks to establish scientifically justified product quality specifications aligned with therapeutic intent and patient needs. It enhances process capability by reducing variability through improved process design, comprehensive process understanding, and robust control strategies. Another essential objective is to strengthen operational efficiency across development and commercial manufacturing by applying risk?based and knowledge?based approaches, enabling better decision?making and resource utilization. Additionally, a QMS aims to reinforce the effectiveness of investigations and root cause analyses, thereby supporting the prevention of recurring issues and ensuring a structured approach to post?approval change management. Collectively, these objectives contribute to sustained compliance, continual improvement, and consistent delivery of high?quality pharmaceutical products.
Pharmaceutical Quality System (ICH Q10) Across the Lifecycle
The ICH Q10 Pharmaceutical Quality System (PQS) provides a comprehensive, lifecycle?oriented model that integrates quality principles across four key stages: pharmaceutical development, technology transfer, commercial manufacturing, and product discontinuation. This unified framework ensures that quality is proactively built into products and processes from initial design through ongoing production and eventual withdrawal from the market.
Effective implementation of ICH Q10 requires strong management responsibilities. Senior leadership is expected to establish a clear quality policy, define measurable quality objectives, and provide adequate resources to support the functioning of the PQS. Additional responsibilities include ensuring transparent communication across the organization, maintaining oversight of outsourced activities and suppliers, and conducting periodic management reviews to evaluate process performance, product quality, and overall system effectiveness.
The PQS is built on four foundational elements:
Process Performance and Product Quality Monitoring Systems, which support continuous evaluation of manufacturing consistency and identification of trends.
Corrective and Preventive Action (CAPA) Systems, which ensure systematic investigation, root?cause identification, and implementation of sustainable corrective actions.
Change Management Systems, which enable structured, risk?based assessment and control of modifications throughout the product lifecycle.
Management Review of Process Performance and Product Quality, which facilitates strategic decision?making and supports continual improvement.
Two critical enablers—Knowledge Management and Quality Risk Management—provide the scientific and analytical foundation for effective PQS operation. Knowledge Management ensures systematic collection, organization, and application of product and process knowledge, while Quality Risk Management promotes structured evaluation of risks to product quality and patient safety. Together, these enablers enhance decision?making, support innovation, and reinforce a state of control throughout the product lifecycle.
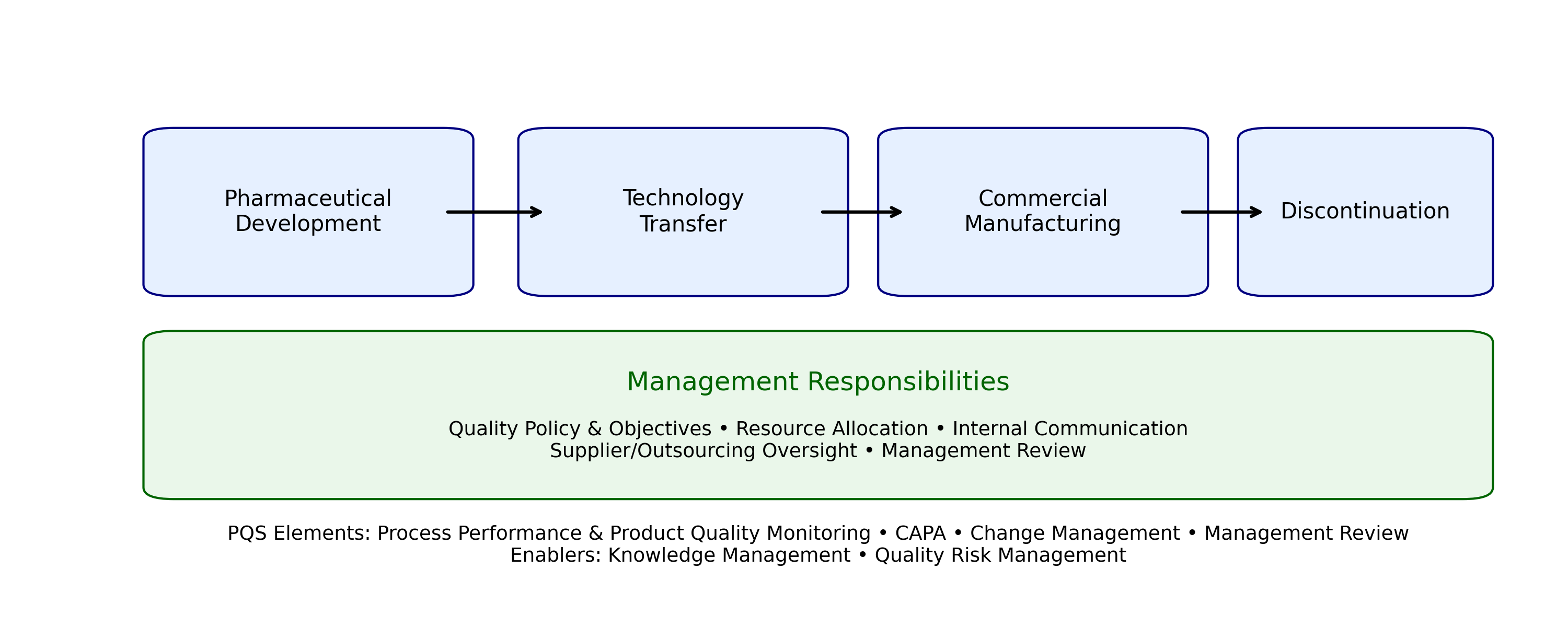
Figure 1. ICH Q10 Pharmaceutical Quality System across the product lifecycle with management responsibilities and enabling elements.
Core QMS Elements and Practical Implementation
Effective functioning of a pharmaceutical Quality Management System (QMS) requires the coordinated implementation of several core elements that collectively sustain a state of control and ensure continual improvement. A key component is Process Performance and Product Quality Monitoring, which involves establishing appropriate leading and lagging indicators, applying statistical and trending tools, and conducting periodic Product Quality Reviews (PQRs) to evaluate manufacturing consistency and detect emerging quality signals. These monitoring systems enable proactive identification of process variability and potential risks to product quality.
The Corrective and Preventive Action (CAPA) system forms another essential pillar of the QMS. It requires comprehensive investigations, rigorous root?cause analysis, and development of effective corrective and preventive measures. CAPA effectiveness checks ensure that implemented actions successfully address the underlying issues, while feedback loops support ongoing knowledge generation and cross?functional learning.
A robust Change Management system is fundamental for controlling modifications across the product lifecycle. Changes must undergo structured, risk?based assessment to determine their potential impact on product quality, regulatory commitments, and process robustness. Cross?functional evaluation, formal approval, implementation planning, and post?change verification ensure that each modification is appropriately justified, executed, and documented.
Finally, Management Review provides strategic oversight of QMS performance. Periodic reviews involve comprehensive assessment of key performance indicators (KPIs), deviation and CAPA trends, complaint and recall data, audit findings, supplier performance metrics, and validation status. These reviews enable leadership to allocate resources effectively, prioritize improvement initiatives, and reinforce a culture of quality throughout the organization.
Change Control Process: Steps And Sequential Flow
An effective Change Control process follows a structured and sequential pathway to ensure that all proposed modifications are scientifically justified, appropriately assessed, and implemented without compromising product quality or regulatory compliance. The process begins with Initiation, where the change is formally proposed and documented with a clear rationale. This is followed by Risk Assessment, performed in alignment with ICH Q9 principles, to evaluate potential impacts on product quality, process performance, regulatory commitments, equipment, facilities, and supply chain continuity. Once risks are assessed, the change undergoes Approval by the relevant cross?functional stakeholders, ensuring that all technical, quality, and regulatory considerations are adequately addressed.
Data Integrity and Documentation Governance (ALCOA+)
Data integrity remains a foundational requirement within pharmaceutical Quality Management Systems, ensuring that all data used for regulatory, quality, and scientific decision?making are trustworthy and reliable. The ALCOA+ principles—Attributable, Legible, Contemporaneous, Original, Accurate, Complete, Consistent, Enduring, and Available—form the globally accepted framework for maintaining data credibility across both paper and electronic systems. Many organizations further extend this model to ALCOA++, incorporating additional expectations such as Traceable and Verifiable, which reinforce the requirement that every data point must be clearly linked to its source, its processing history, and its final use.
Traceable means that data must be contextualized through complete and accurate metadata, audit trails, and system logs that capture who performed the activity, when it was performed, under which conditions, and how it has been processed or modified over time. This includes traceability of raw data to original instruments, traceability of sample identifiers through the workflow, and traceability of analytical results to validated methods and controlled documents. Without traceability, data lose their reliability and cannot support regulatory decisions.
Electronic systems play a critical role in upholding ALCOA++ requirements. They must enforce unique user credentials, role?based access, secure audit trails, electronic signatures, and validated workflows that prevent data overwriting or unauthorized manipulation. Proper governance of metadata—including time stamps, instrument configuration details, audit logs, and file characteristics—is essential to ensure that data remain interpretable long after their creation. Backup integrity, disaster?recovery processes, and archiving controls must preserve both data and metadata in an accessible, tamper?proof manner throughout the defined retention period.
Documentation governance further supports ALCOA++ by requiring controlled templates, formal version control, periodic review cycles, and change control for document revisions. Training records must tie directly to updated documents to ensure personnel follow the most current instructions. In hybrid environments where paper and electronic records coexist, organizations must implement strict procedures for scanning, verification, archival, and retrieval to avoid inconsistencies between formats.
By incorporating Traceable and other enhanced expectations, the ALCOA++ framework strengthens organizational data?integrity culture, reduces compliance risks, and ensures that pharmaceutical decisions are grounded in transparent, reproducible, and scientifically defensible information.
Electronic QMS (eQMS): Transition and Maturity Pathway
The transition from traditional paper?based or hybrid quality systems to an integrated Electronic Quality Management System (eQMS) represents a significant advancement in modern pharmaceutical quality operations. A phased and structured approach to eQMS adoption minimizes operational disruption, reduces manual errors, accelerates audit readiness, and enhances real?time visibility of key quality metrics. This digital transformation enables organizations to strengthen compliance with regulatory expectations while supporting a data?driven culture of continual improvement.An effective eQMS typically incorporates a suite of interconnected modules that automate core quality processes. Document Control forms the foundational module, ensuring standardized templates, version control, electronic approvals, and lifecycle management of controlled documents. The Change Control module streamlines the evaluation, approval, and implementation of changes through risk?based workflows aligned with ICH Q9 principles. CAPA management is enhanced through structured electronic investigation tools, root?cause analysis features, automated reminders for overdue tasks, and integrated effectiveness checks. Deviation and Complaint Handling modules support rapid documentation, investigation, categorization, and closure tracking, reducing the risk of repeated issues.Additional critical components include Training Management, which links competency requirements to current procedures and automatically triggers training assignments following document revisions; Audit Management, which centralizes internal and external audit planning, execution, observations tracking, and follow?up; Risk Assessment tools that enable consistent scoring and mitigation planning; and Equipment or Asset Management modules that maintain calibration, maintenance schedules, and qualification records. Integrated KPI Dashboards provide real?time insights into operational performance, enabling management to monitor trends such as deviation rates, CAPA cycle times, training completion, change?control efficiency, and supplier performance indicators.
The maturity pathway for eQMS implementation typically progresses through several stages. Organizations begin at the Foundational stage, where basic processes are digitized but often function independently. The next stage, Managed, sees cross?functional integration and consistent use of structured workflows. In the Defined stage, standardized processes are fully embedded across departments, supported by validated eQMS modules. The Quantitatively Managed stage introduces real?time metrics, predictive trending, and data?driven risk management. Ultimately, the Optimizing stage reflects a mature eQMS environment where advanced analytics, automation, and continuous improvement practices drive proactive quality management and inspection readiness.Overall, transitioning to a well?designed eQMS enhances transparency, improves data integrity, strengthens regulatory compliance, and enables pharmaceutical organizations to maintain a sustainable state of control across the product lifecycle.
Stages in sequence:
Stage 1: Foundational → Stage 2: Managed → Stage 3: Defined → Stage 4: Quantitatively Managed → Stage 5: Optimizing
The evolution of an Electronic Quality Management System (eQMS) typically follows a structured maturity pathway, progressing through several stages that reflect increasing process standardization, system integration, and data?driven decision?making.
Stage 1: Foundational represents the initial phase, where organizations begin transitioning from paper?based or hybrid systems to basic electronic modules. At this stage, digital tools exist, but workflows may still be inconsistent, and cross?functional integration is limited.
Stage 2: Managed, electronic processes become more uniformly adopted across departments. Standardized procedures emerge, and quality data begin to flow more reliably through defined eQMS workflows. This stage focuses on reducing variability, enforcing compliance, and building user competency with electronic tools.
Stage 3: Defined marks the point where quality processes are fully harmonized and embedded in the organization's operations. End?to?end digital workflows are validated, documentation practices are unified, and cross?functional collaboration becomes more efficient. Electronic modules such as CAPA, change control, and deviations are consistently used, and audit readiness improves significantly.
Stage 4: Quantitatively Managed, organizations leverage real?time dashboards, metrics, and statistical tools to monitor performance. Quality Key Performance Indicators (KPIs) are routinely analyzed to detect trends, predict risks, and guide strategic decisions. Data integrity controls and audit?trail reviews are deeply integrated into routine quality oversight.
Finally, Stage 5: Optimizing represents the highest level of maturity, where advanced analytics, automation, artificial intelligence, and predictive quality models are utilized to proactively manage quality. Continuous improvement becomes embedded in organizational culture, supported by a fully integrated eQMS ecosystem that enhances inspection readiness, operational efficiency, and long?term compliance sustainability.
Table 1. Mapping of PQS Elements to SOPs/Records, eQMS Modules, and KPIs
|
PQS Element |
Typical SOPs/Records |
eQMS Module |
Representative KPIs |
|
Process Performance & Product Quality Monitoring |
Batch records; in-process controls; Product Quality Review; trend reports |
Quality KPIs; Document Control |
First-pass yield; deviation rate/batch; OOS rate; PQR timeliness |
|
CAPA |
Deviation records; investigation reports; CAPA plans; effectiveness checks |
CAPA; Deviations/Complaints |
CAPA on-time closure; effectiveness rate; repeat deviation rate |
|
Change Management |
Change request; impact assessment; approvals; verification protocols |
Change Control; |
On-time change closure; post-change defect rate; retraining compliance |
|
Management Review |
Quality KPIs dashboard; audit summaries; supplier performance reviews |
Quality KPIs; Audits; Supplier Management |
Audit observation closure days; supplier defect rate; training completion |
Supplier and Outsourced Activities
Effective management of suppliers and outsourced activities is a critical component of a robust Pharmaceutical Quality Management System (QMS). Supplier qualification must incorporate a comprehensive, risk?based approach that begins with formal assessment of the supplier’s capability, compliance history, and quality culture. This typically includes risk?based audits, evaluation of manufacturing and quality systems, and verification of adherence to cGMP requirements. Additionally, technical quality agreements must be established to clearly define responsibilities, documentation expectations, communication pathways, deviation handling, and change?control obligations.
Incoming materials—whether active pharmaceutical ingredients (APIs), excipients, packaging components, or critical service outputs—should undergo material controls such as sampling, testing, identity verification, and status labeling to ensure that only conforming materials enter the production process. Ongoing supplier performance monitoring is essential and may include periodic audits, review of on?time delivery, defect rates, complaint trends, Certificate of Analysis (CoA) discrepancies, and responsiveness to investigations.For outsourced GMP activities, such as testing laboratories, contract manufacturers, or service providers, quality agreements must additionally address data?integrity expectations, including ALCOA++ principles, audit?trail review requirements, electronic systems governance, and secure data?transfer controls. Oversight mechanisms must ensure that outsourced partners notify the sponsor of any critical deviations, regulatory inspections, significant observations, or proposed changes through established change?notification pathways. These controls protect the state of compliance, ensure continuity of product quality, and maintain traceability across the extended supply chain.
Quality Key Performance Indicators (KPIs)
1. Deviation Rate per Batch
What it means: Number of deviations recorded divided by total batches manufactured.
Why it matters: Tracks process robustness, identifies recurring issues, and highlights areas needing preventive action.
2. CAPA Cycle Time & CAPA Effectiveness
3. First-Pass Yield (FPY)
What it means: Percentage of batches that meet specifications without rework or reprocessing.
Why it matters: High FPY indicates controlled processes; low FPY signals variability and waste.
4. On-Time Change Closure
What it means: Percentage of change controls closed within the approved timeline.
Why it matters: Reflects Change Management efficiency and regulatory compliance.
5. Complaint Rate per Million Units (PPM)
What it means: Number of market complaints per million units shipped.
Why it matters: A direct measure of product quality and customer experience.
6. Audit Observation Closure Time
What it means: Time taken to close observations from internal/external audits.
Why it matters: Demonstrates responsiveness and preparedness for regulatory inspections.
7. Supplier Defect Rate
What it means: Percentage of incoming material lots rejected or found defective.
Why it matters: Helps monitor supplier performance and raw material quality.
8. Training Completion Timeliness
What it means: Percentage of training completed within due date, especially for SOP revisions and compliance modules.
Why it matters: Ensures personnel qualification and minimizes non?compliance risk.
9. Product Quality Review (PQR/APQR) Adherence
What it means: On?time completion of annual quality reviews for each product.
Why it matters: Shows QMS maturity and compliance with regulatory requirements.
CONCLUSION
An effective pharmaceutical Quality Management System (QMS) harmonizes regulatory foundations with the lifecycle principles outlined in ICH Q10. By embedding risk?based and knowledge?based decision-making across all operations, organizations ensure that quality is built into processes rather than inspected at the end. Robust validation of computerized systems further supports data integrity, traceability, and reliable decision-making.Sustained quality excellence requires disciplined execution of CAPA processes, formalized and controlled change management, and the digital transformation of documentation to minimize human error and improve traceability. Routine monitoring of quality performance indicators and periodic management reviews help maintain a continuous state of control. Organizations that proactively integrate these practices are better positioned for consistent compliance, operational efficiency, and inspection readiness.
REFERENCES

Deepjyoti Das, Quality Management System in Pharmaceuticals: Principles, Practice, and Modernization, Int. J. of Pharm. Sci., 2026, Vol 4, Issue 1, 3563-3571. https://doi.org/10.5281/zenodo.18441253
 10.5281/zenodo.18441253
10.5281/zenodo.18441253