
We use cookies to ensure our website works properly and to personalise your experience. Cookies policy
Clinical pharmacy Nirmal hospital Pvt. Ltd and Maliba pharmacy College Bardoli.
Introduction-Wilson disease, or hepatolenticular degeneration, is a rare autosomal recessive disorder caused by mutations in the ATP7B gene, leading to copper accumulation in various organs, particularly the liver and brain. The clinical presentation is highly variable, often involving hepatic, neurological, and psychiatric symptoms, making diagnosis challenging. Early recognition and treatment are crucial to prevent irreversible organ damage. This review and case report aims to highlight the pathophysiology, diagnostic challenges and importance of early intervention in a paediatric case of Wilson disease. Case Presentation-A 10-year-old boy presented with symptoms including vomiting, abdominal pain, decreased urine output, and weight loss over the preceding two months. He also reported greying of hair. On examination, hepatomegaly and moderate dehydration were noted. Laboratory tests revealed electrolyte imbalances, elevated liver enzymes (SGPT, SGOT), increased ammonia levels, and low ceruloplasmin, suggestive of hepatic dysfunction. Imaging studies confirmed hepatomegaly and fatty liver changes. The diagnosis of Wilson disease was made based on these findings. The patient was treated with copper chelation (D-penicillamine), zinc acetate to reduce copper absorption, and supportive therapies for liver function and ammonia reduction. The patient showed significant clinical improvement during hospitalization and was discharged stable.
Wilson disease, commonly known as hepatolenticular degeneration (HLD) is an autosomal-recessive, monogenic disease. It results from ATP7B (ATPase Activity 7 distinct domain and B class) gene mutations [1]. ATP7B is particularly expressed abundant in the liver and has lesser amounts in the kidney, placenta, brain, lung, and heart. [17] The ATP7B gene is situated on chromosome 13 [5]. There are around 800 distinct ATP7B gene mutations known. [7] This gene encodes the copper-transporting ATPase which helps in transport of copper across the hepatocytes. A typical, balanced diet provides 1.2-1.3mg of copper per day. from which the upper part of the small intestine absorbs about 0.8 milligrams per day. [23] The copper transporters play critical roles in intestinal pathway (ATP7A) as well as hepatic pathway (ATP7B). To enable transport across the apical membrane of enterocytes, dietary cupric copper (Cu2+
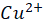
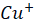

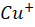
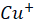
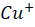

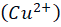
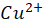
) then binds to α2-macroglobulin (A2M), albumin (ABM) or other minor ligands for delivery to the liver in the portal blood Circulation. [23] This is how copper is transported through the intestine. In hepatocytes the high affinity copper transport protein (CTR1) from the apical side starts the uptake of copper. After entry into hepatocytes copper is distributed to different intracellular compartments such as mitochondria, nucleus and cytosol. [9] Copper attaches itself to the chaperon ATOX1 in the cytosol, which then transfers copper to ATP7B for transit into the trans-Golgi network (TGN) for incorporation into apoceruloplasmin which has eight copper binding sites further it is converted in to ceruloplasmin at the basolateral side of hepatocytes and then released into the systemic circulation. [4][5][8] Mutations in ATP7B specifically missense and frameshift mutations which are associated with severe phenotype of Wilson disease resulting in inadequate copper incorporation into Apoceruloplasmin resulting in reduced ceruloplasmin formation and reduced copper excretion into the bile. [4][5][6] Copper accumulation in hepatic mitochondria induces oxidative stress through fenton reaction, early oxidative aging of DNA, resulting in somatic mutations in the mitochondrial genome. Excess copper in cells can stimulate hydroxyl radical which can damage lipids, proteins and nucleic acids. [10] [11] further it causes hepatocyte damage, inflammation, and fibrogenesis.[3] The gradual buildup of copper in the liver of people with Wilson’s disease starts to surpass the liver’s capacity to store copper and results in damage, most likely as early as age three. Hepatocytes with excess copper promote ATP7B trafficking. The primary homeostatic mechanism for eliminating excess copper from the body is the copper-induced trafficking of ATP7B. [17] Non-ceruloplasmin copper is engulfed by lysosomes and secreted into bile via exocytosis with the help of ATP7B at the canalicular membrane. Copper that has not been transferred in apoceruloplasmin is often held in the cytosol within metallothionein. [8] Excess copper in liver cells remaining after above handling mechanisms will give rise to rupture of hepatocytes and release of excess copper into the blood stream. About 50% of individuals may experience liver impairment that manifests clinically, usually in the middle to late adolescence. [12] Liver disease patients might show up in one of three ways. One is hepatitis, which can present with or without jaundice and increased blood transaminase enzymes. The second is acute hepatic failure, whereas the third is persistent cirrhosis. About 50% of patients have liver injury, including the development of underlying cirrhosis. Copper builds up in the body’s other organs, with the brain appearing to be the next most vulnerable organ. [13] Neurologic ailments can show up as tremor, muscle rigidity, speech problems, gait and balance issues, drooling, difficulty swallowing, and movement disorders. [14][15][16] At the time of diagnosis, the most prevalent clinical signs of Wilson disease were mixed neurologic/psychiatric and hepatic symptoms followed by neurologic/psychiatric, hepatic, and asymptomatic. [14] The prevalence of Wilson’s disease is reported to be between 1/100,000 to 1/3 million individuals. [2] The global incidence of Wilson’s disease is approximately 750000 cases, with the heterozygote carrier rate estimated to be 1 in every 100 individuals. [2] Early diagnosis and treatment are critical in preventing irreversible damage to affected organs. This report discusses a paediatric case of Wilson disease diagnosed through clinical findings, laboratory tests, and imaging studies.
Table 1 A Review of The Role of Copper in Human Health [25][26][27][28]
|
Category |
Details |
|
Foods Rich in Copper |
Copper-rich foods include: |
|
|
1. Shellfish (e.g., oysters, crabs, lobster) |
|
|
2. Organ meats (liver, kidneys) |
|
|
3. Nuts and seeds (e.g., sunflower seeds, almonds, cashews) |
|
|
4. Dark chocolate |
|
|
5. Legumes (e.g., lentils, chickpeas, beans) |
|
|
6. Whole grains (e.g., quinoa, oats, barley) |
|
|
7. Leafy greens (e.g., spinach, kale) |
|
|
8. Potatoes |
|
|
9. Tofu |
|
Recommended Dietary Allowance (RDA) of copper according to National Institute of Health |
Copper RDA (based on age): |
|
|
- Infants: 0–6 months: 200 mcg/day; 7–12 months: 220 mcg/day |
|
|
- Children: 1–3 years: 340 mcg/day; 4–8 years: 440 mcg/day; 9–13 years: 700 mcg/day |
|
|
- Adolescents: 14–18 years: 890 mcg/day (boys and girls) |
|
|
- Adults: 19 years and older: 900 mcg/day (both men and women) |
|
|
- Pregnant women: 1,000 mcg/day |
|
|
- Lactating women: 1,000 mcg/day |
|
Therapeutic Salts for Copper Deficiency |
1. Copper sulphate |
|
|
2. Copper gluconate |
|
|
3. Copper chloride |
|
|
4. Copper ascorbate |
|
Diseases Related to Copper Imbalance |
1. Wilson's disease: A genetic disorder causing copper buildup in organs, leading to liver and neurological damage. |
|
|
2. Menkes disease: A rare genetic disorder causing copper deficiency, leading to neurological and developmental problems. |
|
|
3. Neurodegeneration with Brain Iron Accumulation (NBIA) : A group of rare genetic disorders, some of which may be associated with copper metabolism problems (e.g., Pantothenate Kinase-Associated Neurodegeneration, PKAN). |
|
|
4. Copper toxicity: Caused by excessive copper intake, leading to liver damage, kidney dysfunction, and neurological symptoms. |
Case Presentation
A 10-year-old boy arrived to the hospital with a history of 8–9 vomiting episodes that lasted 2–3 days, along with 1 day of abdominal pain, 1 day of decreased urine output, and 1 day of decreased oral intake. Over the last two months, he has lost 8.5 kg of weight. He also had greying of hair for the past two and a half years. Upon assessment, the patient’s temperature was 98°F, heart rate was 110 beats per minute, blood pressure was 128/70 mmHg, respiration rate was 26 breaths per minute, and SPO2 on room air was 96%. The patient was also conscious and oriented. He had sunken eyes and dry oral mucosa, which were symptoms of moderate dehydration. Hepatomegaly was discovered during the abdominal examination, with a liver spread of +3 cm. The patient's medical history and clinical data, indicated diagnosis of Wilson disease, necessitating further examination.
Table 2 Laboratory Results
|
Parameter |
07/10/2024 |
08/10/2024 |
09/10/2024 |
10/10/2024 |
Normal range |
|
Serum Sodium |
123 ↓ |
122 ↓ |
124 ↓ |
126 ↓ |
135–145 µmol/L |
|
Serum Potassium |
4.6 |
4.7 |
3.5 |
2.9 ↓ |
3.5–5.0 µmol/L |
|
Serum Chloride |
85 ↓ |
84 ↓ |
86 ↓ |
88 ↓ |
98–106 µmol/L |
|
Serum Ammonia |
152↑ |
- |
139 ↑ |
- |
11–35 µmol/L |
|
Creatinine |
0.9 |
0.9 |
0.9 |
0.5 |
0.3–0.7 mg/dL |
|
SGPT/ALT |
192 ↑ |
148 ↑ |
207 ↑ |
133 ↑ |
10–40 U/L |
|
SGOT/AST |
44 |
- |
47 |
133 ↑ |
10–40 U/L |
|
Bilirubin Total |
2.2↑ |
- |
1.4 ↑ |
- |
0.1–1.0 mg/dL |
|
PT/INR |
1.54↑ |
- |
1.10 |
1.15 |
0.9–1.1 seconds |
|
Ceruloplasmin |
- |
- |
14.3 ↓ |
14.2 ↓ |
20–60 mg/dL |
|
Urinary Copper |
- |
- |
189 ↑ |
- |
10–40 mcg/day |
|
Cholesterol |
- |
235↑ |
- |
- |
<170 mg/dL |
|
Triglyceride |
- |
123↑ |
- |
- |
40–130 mg/dL |
|
HDL Cholesterol |
- |
62 |
- |
- |
>45 mg/dL |
|
LDL Cholesterol |
- |
128↑ |
- |
- |
<110 mg/dL |
This table highlights the patient's electrolyte imbalance, liver function abnormalities, elevated ammonia levels, low ceruloplasmin levels and abnormal lipid profile further Imaging studies including ultrasonography of the abdomen, showed moderate hepatomegaly and increased echopattern, suggesting fatty liver changes which aided in the diagnosis of Wilson disease and treatment planning. Laboratory findings clearly suggest Wilson disease-associated Fanconi syndrome, which is characterised by decreased renal tubular function. Persistent low serum Sodium and chloride levels indicate proximal renal tubular failure, which fail to reabsorb filtered sodium and chloride. Hypokalaemia suggests renal potassium wasting, which is a hallmark of proximal tubular failure in Fanconi syndrome. The patient was admitted and started on intravenous fluids due to hyponatremia. 5% NaCl was given as a stat dose, followed by a continuous infusion. IV Vitamin K (10mg/ml) 1ml was administered for three days due to an altered coagulation profile (elevated PT/INR). The patient was also started on capsule D-penicillamine (250 mg) once a day for copper chelation, and zinc acetate(50mg) thrice a day was initiated to reduce intestinal copper absorption. Lactulose was used to reduce ammonia levels, and Aluminium Hydroxide Magnesium Simethicone Gel (gasogel) 10ml twice a day was administered for acidity. The patient's condition improved with supportive management, and the patient was stable at the time of discharge. Over the course of hospitalization, the patient was stabilized, with improvement in electrolyte levels and clinical symptoms. On the 7th day of admission, the patient was deemed stable and ready for discharge. The patient's medications were adjusted, and he was advised to continue D-penicillamine, zinc acetate, and other supportive medications as part of ongoing management.
DISCUSSION
Wilson disease is a relatively uncommon but treatable disorder when detected early. The presentation varies greatly, but common symptoms include hepatic involvement, neuropsychiatric problems, and corneal abnormalities. This patient presented the typical manifestations of Wilson disease, including hepatic dysfunction (increased liver enzymes, hepatomegaly), an abnormal lipid profile indicating hepatocyte damage, this manifests as low serum ceruloplasmin, and elevated ammonia levels, all of which were compatible with the diagnosis. [18] The Fanconi syndrome in Wilson disease results from the toxic accumulation of copper in the renal tubular cells, leading to proximal tubular dysfunction which is seen in this case as electrolyte imbalance. Additionally, some unusual symptoms were observed, such as unintended weight loss and greying of the hairs. These can be brought on by the liver's inability to process and eliminate excess copper, which damages the liver and can cause loss of appetite. Also, as the body's metabolic processes are disturbed, unintended weight loss may result. [19] The oxidative stress brought on by copper buildup can impact several tissues, including hair follicles, even though greying of the hair is not one of the main symptoms of Wilson disease. Furthermore, Wilson disease-related metabolic abnormalities and dietary deficits can accelerate the onset of greying of hair. [20] The elevated ammonia levels and altered coagulation profile (high PT/INR) in this patient suggest the presence of liver dysfunction, likely due to copper accumulation. If Wilson disease moves forward to cirrhosis, lipid abnormalities may change to low cholesterol and HDL levels due to decreased synthesis of lipoprotein. In this case elevated LDL and cholesterol does not indicate any serious liver injury. [21] Wilson disease is managed with copper chelation therapy, usually with penicillamine or trientine, and zinc supplementation to limit copper absorption. The combination of these medicines is successful at lowering copper levels and preventing additional organ damage. The most important treatment for Wilson disease is lifetime oral medication and dietary copper restriction. Liver transplantation, which corrects the underlying hepatic abnormality in Wilson disease, is reserved for severe patients and those who do not respond to medication. [22] In this case, the patient's health improved after early intervention, and he was discharged.
Table 3 Recommended Management of Wilson Disease
|
Drug |
Mode of Action |
Side Effects |
Comments |
|
D-Penicillamine |
General chelator, induces renal excretion of copper |
• Fever, rash, proteinuria, lupus-like reaction • Aplastic anemia • Leukopenia •Thrombocytopenia • Nephrotic syndrome • Degenerative changes in skin • Elastosis perforans serpiginosa • Serous retinitis • Hepatotoxicity • Colitis |
Reduce dose for surgery to promote wound healing and during pregnancy |
|
Trientine |
General chelator, induces renal excretion of copper |
• Gastritis • Aplastic anemia (rare) • Sideroblastic anemia • Colitis |
Reduce dose for surgery to promote wound healing and during pregnancy |
|
Zinc |
Metallothionein inducer, blocks intestinal copper absorption |
• Gastritis • Biochemical pancreatitis • Zinc accumulation • Possible changes in immune function |
No dosage reduction for surgery or pregnancy |
CONCLUSION
The present case demonstrates the importance of investigating Wilson disease in Paediatric patients who present with hepatic failure and also uncommon symptoms such as unintentional weight loss and greying of hairs. Early diagnosis and treatment, especially copper chelation therapy, are critical for avoiding irreversible damage to the liver, brain, and other organs. Regular follow-up and ongoing management are required to observe the patient's improvement and avoid additional complications. The patient's clinical improvement following treatment demonstrates the efficacy of current medications to manage Wilson disease.
ACKNOWLEDGEMENT
We would like to sincerely thank Dr. Vijay Shah for his significant supervision and guidance during the manuscript's preparation. His guidance and support were really helpful in creating this manuscript. We also like to express our gratitude to Nirmal Hospital and its hardworking staff for providing the case report and enabling the resources that helped us in publication.
REFERENCES


Chetan Deore, Twinkle Badlani*, Rare Paediatric Manifestations of Wilson’s Disease: A Case Report and Review, Int. J. of Pharm. Sci., 2025, Vol 3, Issue 4, 80-87 https://doi.org/10.5281/zenodo.15318930
 10.5281/zenodo.15318930
10.5281/zenodo.15318930