
We use cookies to ensure our website works properly and to personalise your experience. Cookies policy
Roorkee College of Pharmacy.
Desmopressin acetate (DDAVP) is an oligopeptide used to treat primary nocturnal enuresis, for example. The low oral bioavailability of DDAVP has expedited the move to other modes of administration such as nasal and oromucosal, with nasal administration resulting in large variations, raising the likelihood of unpleasant side effects. Hyponatremia is common in clinical practice and is mainly caused by renal water retention. Many drugs are thought to be among the different causes of hyponatremia because they either promote the production of arginine vasopressin (AVP) or increase its effect in the kidney. Antidepressants, anticonvulsants, antipsychotics, diuretics, and cytotoxic agents are the most common causes of drug-induced hyponatremia. However, research into the ability of these medicines to boost AVP release from the posterior pituitary gland or raise urine concentration via intrarenal processes is limited. We previously reported that in the absence of AVP, sertraline, carbamazepine, haloperidol, and cyclophosphamide enhanced vasopressin V2 receptor (V2R) mRNA and aquaporin-2 (AQP2) protein and mRNA expression in primary cultured inner medullary collecting duct cells. Tolvaptan, a V2R antagonist, and protein kinase A (PKA) inhibitors both prevented AQP2 upregulation. These observations lead us to the conclusion that the nephrogenic syndrome of inappropriate antidiuresis (NSIAD) is the primary cause of drug-induced hyponatremia. Previous research has also found that the V2R has a role in chlorpropamide-induced hyponatremia. Several additional drugs, such as metformin and statins, have been shown in animal studies to elicit antidiuresis and AQP2 over expression via diverse V2R-independent routes, but they are not related with hyponatremia while being widely used therapeutically
Hyponatremia (serum sodium content <135 mmol/L) is caused by an excess of water in the extracellular fluid. Although sodium depletion often precedes water retention, primary water excess can occur regardless of sodium balance . Water is retained in the body due to excessive consumption and/or decreased renal output. Primary polydipsia is a condition that typically affects neuropsychiatric individuals. The latter can be caused by an absolute reduction in glomerular filtration rate (i.e., kidney failure) or excessively high-water reabsorption along the renal tubule.
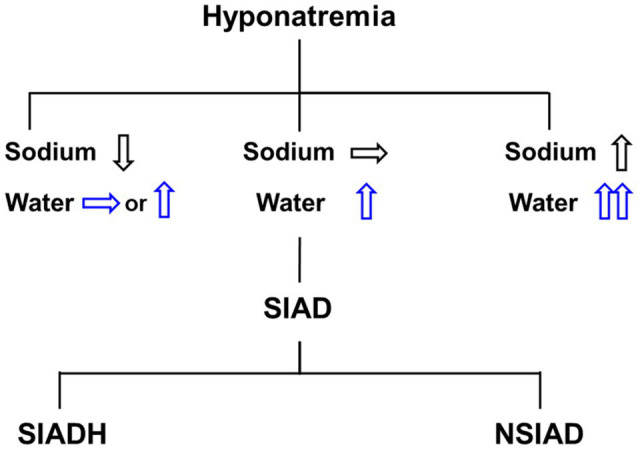
Figure-1
Differential diagnosis of hyponatremia. Hyponatremia can be accompanied by a water and salt balance imbalance, and pure water excess is clinically diagnosed as euvolemic hyponatremia. The most prevalent cause of euvolemic hyponatremia is the syndrome of inappropriate antidiuretic hormone secretion (SIADH), also known as the nephrogenic syndrome of inappropriate antidiuresis (NSIAD). The posterior pituitary gland stores and releases arginine vasopressin (AVP), which regulates renal water excretion. AVP synthesis in the hypothalamus is stimulated by both osmotic and non-osmotic stimuli, including acute systemic hemodynamic alterations, stress, and hypoxia (Schrier et al., 1979). The loop of Henle and collecting duct are the primary sites of AVP action in the kidney. AVP binds to the vasopressin V2 receptor (V2R) at the basolateral membrane of the collecting duct principal cells and induces osmotic water reabsorption by regulating aquaporin-2 (AQP2) and aquaporin-3 (AQP3) water channel proteins (Jung and Kwon, 2016). The thick ascending limb of the loop of Henle is critical in countercurrent multiplication for urine concentration and AVP strongly upregulates the expression of the Na-K-2Cl cotransporter 2 (NKCC2) of the thick ascending limb (Kim et al., 1999). The resultant outer medullary interstitial hypertonicity promotes osmotic water reabsorption along the collecting duct. On the other hand, inner medullary interstitial hypertonicity can be established by urea accumulation facilitated by urea transporters (UTs). By stimulating UT-A1 and UT-A3 in the inner medullary collecting duct and UT-A2 in the thin descending limb of Henle’s loop (Wade et al., 2000), AVP promotes urea recycling. Desmopressin acetate (DDAVP, 1-desamino-8-D-arginin-vasopressin) is a synthetic oligopeptide derivative of the endogenous hormone vasopressin that is used to treat enuresis nocturna and diabetes insipidus centralis in children and adults (De Waele et al., 2014; Lottmann and Alova, 2007). It is also authorized to treat hemophilia A and von Willebrand disease type I (Mannucci et al., 1977). However, orally administered DDAVP is destroyed by the acidic stomach contents, resulting in a low oral bioavailability of 0.08% to 0.16% (Hashim and Abrams, 2008). As a result, parenteral medication administration remains the gold standard for administering peptide-based medicines like DDAVP (Richard, 2017). However, parenteral administration is cumbersome and may lead to low compliance. Various studies are being done to investigate alternate application methods, such as nasal or transoromucosal administration, which have grown in importance in recent years (Patel et al. 2011). Desmopressin is available in a variety of dose forms, including tablet, nasal spray, nasal drop, and, beginning in 2007, sublingual tablet (Vande Walle et al., 2007). By avoiding the intestinal first-pass impact, desmopressin administered via the nasal (5%-10%) and sublingual routes (0.28%) may have higher bioavailability (BA) than oral treatment (Fjellestad-Paulsen et al., 1993; Hashim and Abrams, 2008).
Aim And Objectives
The study's goal was to develop a novel composite dosage form (solid matrix coupled to a bilayer mucoadhesive film) to make DDAVP available orally, lowering the risk of unwanted side effects through precision dosing. DDAVP was integrated into a solid matrix in the form of a minitablet, and both direct tableting (AV > 30) and granulation followed by tableting (AV = 17.86) were evaluated. Granulation and loss supplementing (AV = 11.27) produced minitablets with homogenous content, resulting in prompt drug release (>80% after 7-8 min) and fast disintegration (<49 s). Permeation experiments were conducted with a therapeutically relevant dosage (200 μg) in a time span of up to one hour, resulting in apparent permeation coefficients of 4.90 × 10−6 cm/s (minitablet) and 2.04 × 10−6 cm/s (composite). Comparable fluctuations showed no inferiority of composite and minitablets regarding dosing accuracy. Thus, a step towards controlled and dose-accurate transmucosal delivery of systemically active DDAVP could be achieved.
Objectives-
Drug Profile & Excipients
Antidiuretic Drugs-
An antidiuretic is a drug that regulates fluid balance in an animal's body by lowering urination and countering diuresis.[2] Its effects are opposite those of a diuretic. The main endogenous antidiuretics are antidiuretic hormone (ADH, commonly known as vasopressin) and oxytocin. Both are also used exogenously as drugs in persons whose systems require additional fluid balance support through diuresis suppression. There are also a variety of different antidiuretic medications, some of which are structurally similar to ADH or oxytocin and others not. Antidiuretics diminish urine volume, notably in diabetes insipidus (DI), one of its primary indications. Antidiuretic hormones include vasopressin (ADH), argipressin, desmopressin, lypressin, ornipressin, oxytocin, and terlipressin. Other medications include chlorpropamide and carbamazepine.
Medicinal uses- Diuretics are used to treat heart failure, liver cirrhosis, hypertension, influenza, water poisoning, and a variety of renal illnesses. Some diuretics, such as acetazolamide, serve to alkalize the urine and aid in the elimination of drugs like aspirin in situations of overdose or poisoning. People with eating disorders, particularly those with bulimia nervosa, may overuse diuretics in attempt to lose weight.
Desmopressin and Vasopressin Drugs-
Desmopressin (dDAVP), a synthetic analogue of 8-arginine vasopressin (ADH), is an antidiuretic peptide medication created by deaminating 1-cysteine and replacing 8-L-arginine with 8-D-arginine. ADH is an endogenous pituitary hormone that regulates the body's water content. When ADH is released in response to increased plasma osmolarity or decreased circulating blood volume, it primarily acts on cells in the distal region of the nephron and the collecting tubules of the kidney 6. The hormone interacts with V1, V2, and V3 receptors via different signal cascade mechanisms.
Vasopressin (arginine-vasopressin or antidiuretic hormone) is a nonapeptide generated largely in the brain that performs a variety of physiological tasks relating to diuresis, hemodynamic regulation, and behavior.1,2,3,4,6 Vasopressin is extremely similar to oxytocin, but differs in the third and eighth amino acids.7 Exogenous vasopressin has a number of purposes, but it is most commonly employed to lower blood pressure during systemic shock by enhancing vasoconstriction and renal fluid reuptake via V1 and V2 cellular receptors.6,7,8,10
Excipients- Microcrystalline cellulose(107) Citric acid Monohydrate(109) Carmellose sodiumEthyl cellulose.
Classification of antidiuretic drugs-
1. Antidiuretic Hormone (ADH/Vasopressin) and its Analogs:
2. Thiazide Diuretics:
3. Miscellaneous Drugs:
MATERIALS AND METHOD
Materials-
Antidirutic drugs like as Desmopressin and Vasopressin Samples were acquired as gifts from Indoco Remedies and Ipca Laboratories. Ingredients acquired from Research-Lab Fine Chem Industries include microcrystalline cellulose (Avicel PH 101), PEG 2000, talc, mannitol, lactose, povidone, SSG, and magnesium stearate was received as a kind of gift sample from Evonik Pharma, Mumbai.
Methods-
Drug Excipient Compatibility Study-
Drug-excipient interactions have a significant impact on product performance, both during formulation development and shelf life.16 The compatibility samples were stored in open, parafilm-covered vials with perforations at 40°C and 75% relative humidity for 15 days. The samples were a combination of medication and excipients. Visual and physicochemical inspections were conducted on the items.
Preparation of Sustained Release Pellets-
Pellets were created using the extrusion spheronization method.17 Each weighted batch of excipients and medicine was passed through a No. 40 sieve. To guarantee uniformity, the drug and excipients were thoroughly mixed using a V-cone blender. The correct drug dosage was gradually included into the mixture, resulting in a dough (see Table 1). The extruder made the dough with previously adjusted parameters. The extrudates were collected and placed on the checker plate for spheronization. Spheronization occurred at a specified rate during the ideal time. The spheronizer pellets are dried in an air-heated oven at 80°C for 60 minutes.
Enteric Coating of Sustained Release Pellets by Fluidized Bed Processor-
The fluid bed processor (APCG-225) was loaded with 200 g of weighed drug pellets and warmed for 5 minutes. The pellets were coated with a 3% enteric solution containing isopropyl alcohol from Eudragit S 100 and L 100 55 (in a 1:1 ratio) and 5% triethyl citrate.18 Several factors were used throughout the coating process, including spray gun nozzle size of 1.22 mm, atomizing air pressure of 2.5-0.5 kg/cm2, intake air temperature of 50-55°C, product bed temperature of 40-45°C, and spray rate of 25-50 mL/min. The spray suspension was applied to pellets at a thickness of 8.68% (w/w) of the coating load. It took one hour to coat everything. The coated pellets were dried for one hour at 50°C bed temperature. Pellets were sieved to ensure uniform size distribution.
Compression of Pellets into a Tablet-
Evaluation Of Pellets-
Particle Characterization-
Microscopy was used to measure particle size in several batches of pellet. To analyze the microspheres, a microscope was used to view the slide. The diameter of the pellets indicated their normal particle size.
Flow Properties:
Pellet flow ability was assessed using Carr's Compressibility Index and Hausner Ratio. These are measurements of a powder's compressibility. They were determined using equations 22.
Compressibility Index = 100* [(Vo – Vt )/ Vo]
Hausner Ratio = Vo / Vt.
Angle of repose:
The static angle of repose was computed using both the fixed funnel and free-standing cone approaches. A funnel with a tip 2 cm above a blank sheet of paper was put on a flat, horizontal surface and fixed to a retort stand. till the top of the cone. Once the powder had reached the top, it was gently poured down the funnel. Following four measures, The average base diameter of the powder cone was determined and an equation was used to compute the angle of repose.22
θ = tan-1 (2h/D)
h = height of the powder cone
D = diameter of the cone’s base
Friability:
The combined impacts of abrasion and shock were applied to Ten grams pellets in a friabilator. For 4 minutes, the pellets were vibrated at 100 rpm.23
Assay:
The crushed pellets had been accurately weighed. The gliclazide powder (80 mg) was placed in a 100 mL volumetric flask. A volumetric flask of methanol was placed inside a sonicator for 15 minutes. 100 mL was added to the capacity. After filtering the solution, remove 1-mL of the filtrate and dilute it to 100 mL. The absorbance of this solution was determined and documented. To quantify the amount of medication in the weighted pellets, a linearity equation was used based on a calibration curve.
Sieve Analysis:
It measures the distribution of aggregate particle sizes in a sample. The sieves used were 16#, 20#, 30#, and 100#, with a pan at the bottom to capture particles. The sieve tower was shaken for 10 minutes in ascending sequence of sieve number (16# at the top to 100# at the bottom). After 10 minutes, particles remained on each filter and were weighed.24
RESULTS AND DISCUSSION
Dissolution Studies:
Dose dumping is a major issue with continuous-release dose formulations. In vitro dosage dumping relies heavily on the early release of active ingredients. The European Medicines Agency (EMEA) sets a limit of 20-30%.28 The study aims to develop a bioavailability extended-release pharmaceutical dose form that improves patient compliance while releasing no more than 30% of the active ingredient in less than two hours. Dissolution experiments on the pellets showed delayed release behavior. Within the first two hours of the optimized batch F8's release of NMT 30, 98.2% of the active component was released. The pellets' covering layer stayed intact. However, there was considerable swelling during the release surgery. The polymer and coating release ratios significantly effect the release. The inclusion of rate-delaying polymers (MCC) in core pellets and Eudragit S 100 and L 100 in coating resulted in extended release delivery.29 In all cases, "n" in the Korsmeyer-Peppas equation was more than 0.89. This shows that the medication was released mostly through relaxing, similar to how polymers expand in water. Polymer erosion is an additional phase in this process.30
Compressed Tablet Evaluation:
The tablets were evaluated for their hardness, friability, drug content, weight uniformity, thickness, and in vitro dissolution. The tablets maintained their spherical form and had no visible cracks. Each tablet has a similar thickness. The thickness range was 3.18-3.76 mm. Official criteria indicate that all pill formulations passed the weight uniformity test, with average percentage deviations falling within the permitted range. Across many batches of tablets, the quantity of medication was consistently higher than 90%. The tablet formulations exhibited a hardness of 4 to 5 kg/cm2. The firmness of a tablet cannot be determined just by its hardness. Friability is another metric for assessing tablet strength. Conventionally compressed tablets are generally regarded as appropriate if they lose less than 1% of their weight. The percent brittleness in the current study was less than 1% for all formulations, showing that the brittleness was within the permitted limits.30 Table 6 displays the physicochemical evaluation of compressed tablets.
REFERENCES


Shweta Saini*, Amit Kumar, Sustained Release Pellets for Antidiuretic Drugs Formulation & Clinical Evaluation, Int. J. of Pharm. Sci., 2025, Vol 3, Issue 4, 3131-3138 https://doi.org/10.5281/zenodo.15295146
 10.5281/zenodo.15295146
10.5281/zenodo.15295146