
We use cookies to ensure our website works properly and to personalise your experience. Cookies policy
Indira College of Pharmacy, Vishnupuri, Nanded, Maharashtra, India
Personalized medicine (PM) has transformed oncology by tailoring chemotherapeutic interventions to the genetic, molecular, and clinical characteristics of individual patients. Within the European Union (EU), the regulatory framework for such innovative therapies is complex, involving coordination between the European Medicines Agency (EMA) and the In Vitro Diagnostic Regulation (IVDR) (EU 2017/746) for companion diagnostics (CDx). This review provides an in-depth overview of the approval pathway for personalized chemotherapy in the EU, covering regulatory mechanisms, clinical trial regulations, and adaptive approval schemes such as PRIME, conditional marketing authorization (CMA), and accelerated assessments. The EMA’s centralized authorization process, combined with the Clinical Trials Regulation (EU No 536/2014), ensures consistent evaluation and transparency across Member States. Ethical, scientific, and logistical challenges—such as data privacy under the General Data Protection R egulation (GDPR), evidence generation for small biomarker-defined populations, and disparities in patient access—remain major hurdles. Comparative insights between EU and US frameworks highlight the global trend toward harmonized, evidence-based regulatory science. Future developments, including the European Health Data Space (EHDS), DARWIN EU®, and artificial intelligence (AI)-driven pharmacovigilance, are expected to streamline the assessment of personalized chemotherapies. Overall, this review emphasizes that the EU’s evolving regulatory ecosystem continues to balance innovation, patient safety, and equitable access in the era of precision oncology.
1.1 Background
According to the World Health Organization (WHO), cancer still accounts for around 10 million deaths annually, making it a major worldwide health concern. Traditional cytotoxic chemotherapy has saved many lives, but significant inter-patient variation in toxicity and efficacy limits its practical usefulness. Inconsistent treatment results are frequently caused by variations in medication metabolism, tumor heterogeneity, and genetic makeup. Personalized (precision) medicine, which matches treatment to each patient's own genetic, genomic, and clinical profile, has been adopted by contemporary oncology to overcome these constraints. A significant shift from standardized treatment plans to evidence-based customization is represented by personalized medicine. Clinicians can choose the best dosage, anticipate therapeutic response, and prevent needless exposure to ineffective medicines by using genetic and biomarker data. Companion diagnostics (CDx), which are validated in vitro assays that can identify individuals most likely to benefit from a targeted medication or at risk of severe side responses, are often used in conjunction with this method in cancer.
A complicated yet unified regulatory framework supports precision oncology innovation throughout the European Union (EU). While diagnostic devices are governed by the In Vitro Diagnostic Regulation (IVDR) (EU) 2017/746, therapeutic items are mainly assessed by the European Medicines Agency (EMA). In order to guarantee analytical and clinical validity, the EMA and authorized notified entities must coordinate the review of a medicinal product's co-development and its diagnostic test . By enabling quicker evaluation of treatments that meet unmet medical needs, recent policy measures like the PRIME (Priority Medicines) program and the Clinical Trials Regulation (EU) No 536/2014 have further modernized the approval environment.
1.2 Concept of personalized medicine
To improve diagnosis and direct treatment selection, personalized medicine combines clinical data with high-dimensional biological data, such as genomic, transcriptomic, proteomic, and metabolomic profiles. Numerous oncogenic mutations that underlie tumor behavior have been discovered by researchers since the Human Genome Project was completed. HER2 amplification in breast cancer and EGFR mutations in non-small-cell lung cancer are two examples. Targeted drugs with better selectivity and tolerability than traditional chemotherapy, like trastuzumab, vemurafenib, and imatinib, have been made possible by these molecular findings.
Three key biomarker categories are included in precision oncology in practice:
Healthcare professionals can optimize efficacy, minimize adverse responses, and more effectively manage resources by integrating these indicators into treatment decision-making. As a result, the more general goal of "the right drug, at the right dose, for the right patient" is supported by customized medicine.
1.3 Personalized medicine in chemotherapy
Systemic toxicity and inconsistent patient benefit result from the indiscriminate cytotoxic effects of traditional chemotherapy on quickly proliferating cells. Clinicians can now choose anticancer medications based on tumor-specific changes rather than just histopathological characteristics thanks to molecular profiling. For example, the detection of BRAF V600E mutations in melanoma directs the use of selective kinase inhibitors like vemurafenib or dabrafenib, while the identification of KRAS mutations in colorectal cancer predicts the lack of effect from anti-EGFR monoclonal antibodies like cetuximab. In order to maximize clinical results, this paradigm—often referred to as customized chemotherapy—combines targeted treatments with molecular diagnostics. These correlations are confirmed by biomarker-driven clinical trials, which show increased response rates, decreased toxicity, and improved survival.
The body of research demonstrates that the therapeutic index of cancer chemotherapy can be greatly increased by customizing cytotoxic or targeted regimens to each patient's unique tumor biology.
2. Methodology:
2.1 Regulatory framework in the European Union
Three main legislative tools form the basis of the European regulatory framework for pharmaceuticals and diagnostics:
2.2 European medicine Agency (EMA)
Through its Committee for Medicinal Products for Human Use (CHMP), the EMA oversees the evaluation of pharmaceuticals. All EU Member States are subject to the decision once a marketing authorization is approved under the centralized process. Comprehensive non-clinical and clinical data are used to assist the evaluation of applications based on quality, safety, and efficacy criteria.
2.3 In Vitro diagnostic regulation (IVDR)
Stricter criteria for analytical and clinical validation of diagnostic instruments are introduced by the IVDR, which replaces Directive 98/79/EC. When associated with a par ticular medication, companion diagnostics are classified as Class C (high-risk) and require both scientific consultation with the EMA and conformity assessment by a notified organization. The adequate demonstration of diagnostic performance for its intended clinical purpose is guaranteed by this dual-review mechanism.
2.4 Clinical Trial Regulation (CTR) (EU NO 536/2014)
Through the Clinical Trials Information System (CTIS), the CTR unifies trial authorization and supervision among Member States . The legislation makes it easier to evaluate biomarker-based oncology trials, which frequently involve tiny, genetically defined populations, by simplifying multi-country applications and increasing transparency.
2.5 Approved Pathway for Personalized Chemotherapy
The sequential pre-clinical, clinical, and post-authorization stages of the regulatory process for customized chemotherapeutic products are coordinated between pharmacological and diagnostic assessments.
2.5.1 pre-clinical phase
Establishing pharmacological rationale and finding biomarkers that correspond with treatment response are the main goals of early development. To verify repeatability, sensitivity, and specificity at this point, analytical validation of related diagnostic assays is essential.
2.5.2 Clinical development
Targeted agent clinical trials are often organized around cohorts specified by biomarkers. As demonstrated by programs like NCI-MATCH and Lung-MAP, novel designs like adaptive, umbrella, and basket trials allow for the concurrent evaluation of several targeted medicines across genetic subgroups. In uncommon molecular subgroups where traditional randomized trials are not feasible, these adaptable approaches facilitate the collection of evidence.
2.5.3 Marketing authorization
Sponsors submit a Marketing Authorization Application (MAA) with information on chemistry, production, non-clinical and clinical findings, and pharmacovigilance systems to the EMA after a successful clinical evaluation. A favorable CHMP review results in the European Commission's final authorization . Concurrently, a notified body independently evaluates the companion diagnostic's compliance. The precise alignment of diagnostic validation with the therapeutic indication listed on the drug's label is ensured by close coordination .
2.5.4 Post-marketing surveillance
Both drugs and diagnostics are subject to ongoing monitoring after approval. Implementing Risk Management Plans (RMPs), post-authorization safety studies (PASS), and continuous performance monitoring for diagnostics are among the responsibilities. Over the course of a product's life, this surveillance framework ensures its performance and safety.
2.6 Accelerated and Conditional Approval Pathway
The EMA has developed a number of regulatory tools that permit flexible evaluation schedules and adaptive evidence gathering in order to enhance patient access to cutting-edge cancer treatments. The PRIME (Priority Medicines) Scheme encourages researchers to produce reliable clinical data effectively by offering early and ongoing regulatory advice for medications that treat conditions with significant unmet needs . When the benefit-risk ratio is favorable but complete clinical data are not yet available, conditional marketing authorization (CMA) allows for temporary approval. After authorization, sponsors are required to provide supporting documentation . For items deemed to be of substantial public health concern, Accelerated Assessment shortens the typical evaluation period from 210 days to 150 days.
Adaptive pathways, which provide iterative evidence generation and tiered approval procedures, are especially helpful for uncommon or biomarker-defined groups when large-scale studies are not feasible . When taken as a whole, these systems offer a methodical way to accelerate the availability of customized cancer treatments, guaranteeing prompt patient access while maintaining regulatory and scientific integrity .
2.7 Ethical and Scientific Challenges
Precision medicine concerns a number of ethical, societal, and scientific issues even though it is a breakthrough in oncology:
Health Technology Assessment (HTA): The rate and uniformity of clinical uptake are impacted by country differences in post-approval reimbursement regulations .
2.8 Case Examples
The following items serve as examples of how the EU approval process incorporates personalized medicine principles:
These illustrations show that a paradigm change toward biomarker-driven approvals is reflected in the growing reliance of regulatory decisions on confirmed molecular diagnostics to identify eligible patient populations
2.9 Future Perspective
Rapid advancements in data science, artificial intelligence, and genomics have led to ongoing changes in the EU regulatory framework. Promoting the use of real-world data (RWD) and real-world evidence (RWE) for clinical and regulatory decision-making is the goal of strategic initiatives like Horizon Europe and the European Health Data Space (EHDS) . Pharmacovigilance and post-authorization research activities are better integrated with RWD thanks to the EMA's DARWIN EU® initiative . It is anticipated that future regulatory science will prioritize interdisciplinary collaboration between the EMA, notified bodies, and HTA agencies. To speed up evaluation, encourage ongoing learning, and cut down on duplication in regulatory procedures, improved digital infrastructure and open data exchange will be essential. In the end, standardizing scientific assessment and ethical supervision will guarantee that advancements in precision chemotherapy result in real advantages for patients across Europe
2.10 Rapid Growth of Personalized Medicine & Complex regulatory Landscape
Molecular biomarkers and companion diagnostics (CDx) are driving the rapid evolution of personalized chemotherapy. The particular difficulties of precision oncology might not be adequately addressed by conventional clinical and regulatory frameworks. Optimizing patient care and safety requires an understanding of how the EU approves such therapies. Personalized treatments are governed by the Clinical Trials Regulation (CTR), the In Vitro Diagnostic Regulation (IVDR), and the European Medicines Agency (EMA). These structures are intricate and often changing. A thorough analysis aids in finding loopholes, streamlining procedures, and enabling prompt approvals.
3. Growth in The Global Personalized market
The global PM market is experiencing remarkable growth, driven by rising demand for customized therapies and precision diagnostics. The market’s current trajectory suggests significant expansion in the next decade, with North America leading the way as of 2023. This growth is attributed to the adoption of advanced healthcare IT systems, artificial intelligence (AI) for drug development and interaction modeling, and next-generation sequencing (NGS) technologies for personalized data generation. However, balancing the high costs of infrastructure with the quality and efficacy of PMs remains a critical concern. Research institutions are playing an increasing role in addressing these challenges, particularly through the integration of pharmacogenomics, which is expected to further drive innovation in R&D for PM. Illustrated in fig. 1.
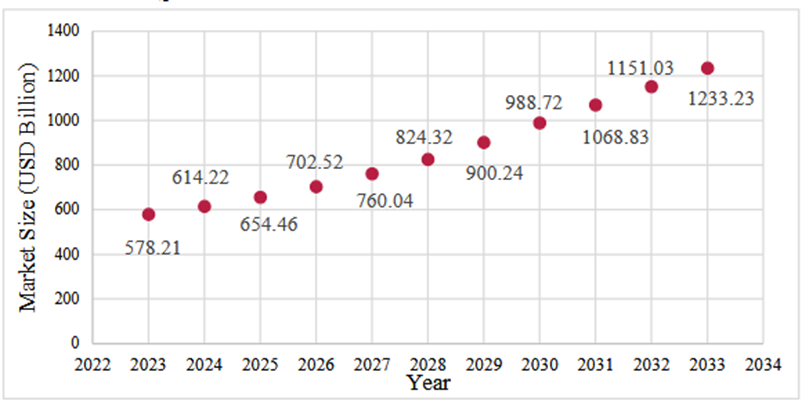
Fig.1: Global Market Size for Personalized Medicine (billion USD)
4. Legislative Policies in the USA & EU for Personalized Medicine
Initiatives like the Critical Path Initiative in the US promote business partnerships to tackle the issues in drug development and accelerate the pace of personalized medicine advancement. As a regulatory agency, the FDA could expedite the development process by endorsing conditional approvals and permitting more affordable and smaller clinical trials for customized medications. Clinical Outcomes of patients and compliance can be tracked via personal Digital Health Technologies (DHTs), ensuring the quality and safety of the treatment [48]. The European Union's governing body, the European Medicines Agency (EMA), is in charge of advancing public health and safety and overseeing and approving innovative treatments and diagnostics. The Centralized approval process of EMA expedites access to PMs across all Member States [49]. Providing patients access to safe and effective medications is one of the EMA's main goals. An "EMA/CHMP think-tank group on innovative drug development" was established to achieve this goal. This specialist group seeks to discover scientific roadblocks in the R&D of the pharmaceutical sector as well as in academic settings that impede the development of novel medications. A comparative analysis of regulations and guidance documents related to personalized medicine in the US and EU is shown in Table 1.
As a next step, cooperation between regulatory bodies, healthcare providers, patient advocacy organizations, and industry players is necessary to develop a legislative framework that promotes innovation, safeguards patient confidentiality, and guarantees the efficient and secure provision of customized medical care. Furthermore, to address the significant disparities in therapeutic accessibility, there is an urgent need for global harmonization across regulatory bodies. To improve accessibility and generalisability, novel therapies should be subjected to Global Clinical Trials (GCTs). Since it is not always practical to design Randomized Controlled Trials (RCTs) involving patients from different countries and ethnicities, it is important to adopt innovative trial designs and complex trials for PMs for rare diseases and other critical disorders.
Table 1: Regulatory Aspects of Personalized Medicines in the US and EU
|
Aspect |
USA |
Europe |
|
Regulatory Body |
US Food and Drug Administration (FDA) |
European Medicines Agency (EMA) |
|
Preferred Term |
Precision Medicine |
Personalized Medicine |
|
Key Documents |
- Public Human Genetic Variant Databases - Design and Validation of NGS-Based IVDs for Germline Disease Diagnosis |
- Regulation (EU) 2017/746 (IVDR) - Regulation (EU) 2016/679 (GDPR) - Regulation (EU) No 536/2014 (Clinical Trials) |
|
Initial Milestone |
Precision Medicine Initiative launched by the US President on December 18, 2015. |
European Commission Conference held in 2011 to explore PM perspectives. |
|
Policy Development |
Critical Path Initiative |
EMA/CHMP Think-Tank Initiative to foster innovative therapies. |
|
Support Structures |
Human Genome Project |
ERA-PerMed (European Research Area Network for Personalized Medicine) |
Table 2: Approved Personalized Medicines in Chemotherapy in 2023 in EU
|
Sr. No. |
Region |
Active Ingredient |
Brand Name |
Indication/ Treatment |
|
1 |
EU |
Nivolumab |
Opdivo |
Melanoma, non-small cell lung cancer |
|
2 |
EU |
Durvalumab |
Imfinzi |
Non-small cell lung cancer |
|
3 |
EU |
Elotuzumab |
Empliciti |
Multiple Myeloma |
|
4 |
EU |
“Pembrolizumab” |
“Keytruda” |
Melanoma: NSCLC: urothelial cancer: head neck cancer |
|
5 |
EU |
Necitumumab |
Portrazza |
Squamous non-small cell lung cancer |
5. Transformational Impact of PM in Oncology
PM has revolutionized oncology, offering transformative advancements in treatment approaches that enhance patients' quality of life. With a deeper understanding of host-related factors, PM ensures long-term survival, reduced toxicity, and improved effectiveness in cancer treatments. For instance, the FDA and the National Comprehensive Cancer Network (NCCN) have established protocols combining chemotherapy with immunotherapies (e.g., ipilimumab and nivolumab) for treating non-small cell lung cancer (NSCLC), achieving high success rates. These innovations are supported by breakthroughs in identifying driver mutations, resistance mutations, and the interplay between cancer and the immune system. However, the implementation of such therapies is often hindered by high costs, requiring innovative solutions to make PM more accessible.
Table 3: FDA issued guidance documents on PM
|
Title |
Year |
|
“IND Submissions for Individualized Antisense Oligonucleotide Drug Products: Administrative and Procedural Recommendations Guidance for Sponsor-Investigators” |
2021 |
|
“Enrichment Strategies for Clinical Trials to Support Determination of Effectiveness of Human Drugs and Biological Products” |
2019 |
|
“Principles for Co-development of an In Vitro Companion Diagnostic Device with a Therapeutic Product” |
2016 |
|
“Clinical Pharmacogenomics: Premarketing Evaluation in Early Phase Clinical Studies and Recommendations for Labelling” |
2012 |
|
“E16 Guidance on Biomarkers Related to Drug or Biotechnology Products Development: Context, Structure, and Format of Qualification Submissions” |
2011 |
|
“In vitro Companion Diagnostic Devices” |
2011 |
|
“Qualification Process for Drug Development Tools Guidance for Industry and FDA Staff” |
2010 |
|
“E15 Definitions for Genomic Biomarkers, Pharmacogenomics, Pharmacogenetics, Genomic Data and Sample Coding Categories” |
2008 |
|
“Pharmacogenomic Tests and Genetic Tests for Heritable Markers” |
2007 |
CONCLUSION
The approval process for personalized chemotherapy in the European Union represents a significant advancement in regulatory science, integrating molecular diagnostics with therapeutic evaluation to deliver individualized treatment. The collaboration between the EMA, notified bodies, and Member State authorities under frameworks such as the IVDR and CTR ensures a harmonized and science-driven approach to precision oncology. Future regulatory evolution will increasingly rely on digital infrastructure, real-world data (RWD), and cross-sector collaboration to support efficient evidence generation and post-marketing surveillance. By maintaining a balance between innovation, safety, and accessibility, the EU continues to set a global benchmark for the governance of personalized medicine in chemotherapy. While substantial progress has been made in streamlining approvals through adaptive and accelerated pathways, persistent challenges such as the synchronization of drug–diagnostic co-development, ethical management of genomic data, and equitable access to targeted therapies across Member States must still be addressed.
CONFLICT OF INTEREST
The authors declare no conflict of interest. This review is based on publicly available regulatory and scientific literature, and no financial or personal relationships have influenced its preparation or conclusions.
REFERENCES


Nikita Delmade, Rohit Muneshwar, Dr. Vijay Navghare, Dr. Suryakant Jadhav, Bhagwat Deshmukh, The Approval Process of Personalized Medicine for Chemotherapy in European Union, Int. J. of Pharm. Sci., 2026, Vol 4, Issue 2, 4183-4192. https://doi.org/10.5281/zenodo.18791705
 10.5281/zenodo.18791705
10.5281/zenodo.18791705