Department Of Pharmaceutics , School Of Pharmacy , Abhilashi University Chailchowk Distt Mandi
Objective: The goal of the current study was to create transdermal patches that would serve as an alternative dosage form for the parenteral proton pump inhibitor (PPI), which is currently used to treat peptic ulcers through oral administration. Methods: Using a solvent evaporation approach, transdermal patches containing PPI were produced using HPMC E5 with PVP K 30 and HPMC E5 with Eudragit L100 polymers in varied ratios. A range of evaluation parameters , including thickness, folding endurance, weight uniformity, content uniformity, swelling index, percentage moisture content, moisture uptake, surface pH, and in vitro release experiments, were applied to all of the constructed patches. Results: In the transdermal patch release studies, formulation F1 demonstrated a prolonged drug release (98.99 %) for 24 hours, indicating the drug's maximum availability. Conclusion: In conclusion, this implies that the regulated release of pantoprazole sodium via transdermal administration is maintained for a prolonged duration.
Transdermal patches are designed to treat a variety of ailments, and the transdermal drug delivery system (TDDS) is a generally recognized method of drug delivery. Transdermal administration improves patient compliance and circumvents the first pass metabolism, which result in over-injectable and oral routes, respectively. They can even stop low absorption and gastrointestinal issues brought on by medications. (1) Maximizing the amount of skin flux into systemic circulation while simultaneously lowering the medication's retention and metabolism in the skin is the aim of the transdermal drug delivery method. These medical advantages demonstrate TDDS's increased marketing potential. . Since the majority of drug molecules enter the skin through the intercellular micro route, permeation or penetration enhancers play a crucial role in TDDS because they can reverse the stratum corneum's barrier resistance without endangering live cells.(2) In 1981, the FDA approved the first transdermal patch, called Transderm-Scop, which was created by Alza Corp. in California to cure motion sickness using the medication scopolamine. Transderm-nitro was then licensed by the FDA to treat angina pectoris. Owing to its on-going success, 35 TDDS patches for a range of conditions, including male hypogonadism, angina pectoris, motion sickness, hypertension, and female menopause, are presently available on the market. Transdermal delivery had a $12.7 billion market share in 2005; by 2010, it had grown to $21.5 billion, $31.5 billion, and was still growing annually.(3) Proton pump inhibitors, such as pantoprazole, are substituted benzoimidazole sulphoxide used to treat gastrointestinal disorders associated with acid reflux, including duodenal and gastric ulcers, reflux esophagitis, and others. When taken as an enteric-coated 40 mg tablet, pantoprazole is absorbed quantitatively. After several doses, its absolute bioavailability remains constant at 77%.(4)The pharmacokinetics of pantoprazole exhibit linearity following intravenous and oral dosing. Because pantoprazole is heavily processed in the liver, the current study attempted to develop a transdermal drug delivery system for it in order to overcome these issues.(5)
MATERIALS AND METHODS:
Materials:
A sample of pantoprazole was received from Dr. Reddy's Laboratories Ltd. in Hyderabad. Sodium hydroxide, PVA, and potassium dihydrogen phosphate were bought from Thomas Baker (Chemicals) Pvt Ltd in Mumbai. The supplier of HPMC E5 was Loba Chemi Pvt Ltd in Mumbai. We bought PVP, Methanol, Dibutyl Phthalate, Chloroform, and DMSO from Research-Lab Fine Chem Industries in Mumbai. The supplier of Eudragit L100 was Rohm Pharma in Germany. The analytical grades applied to all other reagents. (6)
Preparation of backing membrane:
4%w/v polyvinyl alcohol (PVA) was dissolved in water to create an aqueous solution for the backing membrane. After adding 4 grams of PVA to 100 millilitres of warm, distilled water, the mixture was continuously stirred and heated to 60 degrees Celsius for a brief period of time to create a uniform solution. Next, glass Petri plates measuring 63.5 cm2 were filled with 15 ml of the homogeneous solution, and they were left to dry for 6 hours at 60 °C in a hot air oven.(7)
Preparation of placebo films:
Using the hit-and-trial method, different combinations of hydrophilic and hydrophobic polymers were used to generate the various placebo films. The polymeric combinations chosen for the fabrication of drug-incorporated matrix systems showed smooth and flexible films. (8) The solvent evaporation method was used to prepare each film. Using various ratios of Hydroxy Propyl Methyl Cellulose (HPMC E5) with Polyvinylpyrrolidone (PVP), Ethyl cellulose, Eudragit L 100, and Eudragit S100, the matrix-type transdermal patches containing Pantoprazole Sodium were created.
Formulation of transdermal patches:
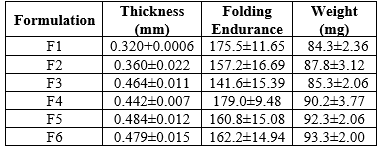
Using various polymers, transdermal films containing pantoprazole sodium were cast on a petri dish using a solvent evaporation technique.(HPMC E5:Eudragit L 100 and HPMC E5:PVP K30). The ratio of drug to polymer was set at 1:1, whereas the ratio of polymer to polymer was set at 1:2, and 2:1. (9) All six formulations included three distinct HPMC E5 concentrations, and each of the other two polymers, PVP K 30 and Eudragit L100, was utilized in each of the three formulations at a different concentration (table 1). Propylene glycol and N-butyl phthalate were employed as plasticizers.(10)
Formulation & Ingredients Table of Pantoprazole Sodium Transdermal Patches(11)
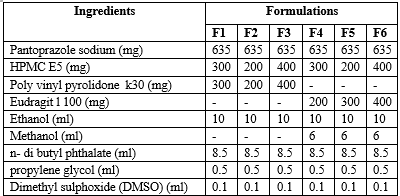
(All quantities are in mg/ml )
In order to create a transparent solution, the polymers were precisely weighed, dissolved in 10 mL of ethanol, and, in the case of Eudragit L 100,chloroform: methanol (1:1) solution was also utilized. The medication pantoprazole sodium was dissolved in the aforementioned mixture and stirred until a clear solution formed. Subsequently, the plasticizer and permeability enhancers were gradually added to the formulation and thoroughly mixed. (9)
After a 24-hour room temperature drying period and glycerine lubrication, the homogeneous solution was transferred to a petri plate. The petri dish was covered with an inverted funnel to stop the solvent from evaporating too quickly. The dried patches were removed after 24 hours and kept in a desiccator for further studies.
Evaluation of transdermal patches
A certain (2 by 2 cm) section of the strip was cut consistently, then folded repeatedly until it broke. The number of times the film was folded at the same spot, either to break the film or to cause visible cracks, defined the folding endurance value.(12)
A tensiometer was used to measure the patch's tensile strength It consists of two load cell grips. While the upper one was movable, the lower one was fixed. The 2x2 cm film strips were positioned in between the cell grips, and force was gradually applied until the film broke. Using the dial reading in kilos, the tensile strength was noted
The percentage elongation break was calculated by noting the length just before the breaking point and the following formula was used to calculate the percentage elongation:(13)
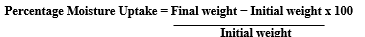
The thickness of the transdermal patches was measured using a digital micro meter screw gauge at three different places, and the mean value is calculated.
A 2x2 cm size transdermal patch was dissolved in 100 ml methanol and shaken continuously for 24 h. The whole solution was then ultra sonicated for 15 min. After filtration, the drug's content was measured using spectrophotometry at a wavelength of 292 nm.(14)
The prepared transdermal films were individually weighed and stored in a desiccator containing a fused saturated solution of potassium chloride. After 24?h, the films were reweighed and the percentage moisture uptake was calculated using the following formula:
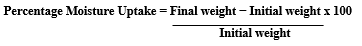
The formulated transdermal patches were weighed (W1) individually and incubated at 37±0.5 ° C separately in agar gel (2%) plate.
The patches were removed from the petri dish at regular time intervals of every 15 min up to 1 h and the excess water on the surface was removed carefully with filter paper. The swollen patches were reweighed (W2) and the swelling index was calculated by using the formula:
Swelling index = W2 ? W1 / W1 x 100(15)
In-vitro drug release studies:
For the in vitro drug release experiments, a Franz diffusion cell with a receptor compartment capacity of 60 ml was employed. Using a cellulose acetate membrane from the transdermal matrix-style patches that were manufactured, the medication was identified. The donor and receptor compartments of the diffusion cell were divided by a cellulose acetate membrane with a pore size of 0.45 ?. The cellulose acetate membrane was covered with the manufactured transdermal patch and sealed with aluminum foil. The receptor compartment of the diffusion cell was filled with 7.4 pH phosphate buffer.(16)
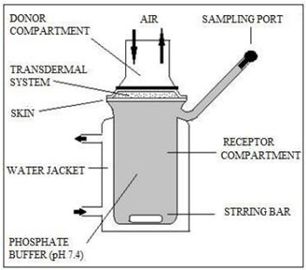
Fig No 1.- Franz diffusion cell
The entire assembly was placed on a hot plate magnetic stirrer, and throughout the experiments, magnetic beads in the receptor compartment were used to continuously stir the solution at 50 rpm while maintaining the temperature at 37±0.5 °C, or the average body temperature. The samples were collected at different times and their drug content was determined using spectrophotometry. Air bubbles can readily enter the receiver compartment during manual sampling, so it is necessary to pay close attention to it the entire time during the experiment. The receptor step was refilled with an equivalent volume of phosphate buffer for each sample removal.(17)
In vitro permeation study:
Male Wistar rats weighing 200–250 g had their full-thickness abdomen skin used for an in vitro permeation research employing Franz diffusion cells. Using an electrical clipper, the hair in the abdomen area was carefully removed. Distilled water was used to completely clean the skin's dermal side in order to eliminate any tissue or blood vessel adhesions.
Before starting the experiment, it was equilibrated in phosphate buffer saline (pH 7) for an hour. The compartment was kept at 37±0.5 °C by means of a heater that was controlled using thermostatic principles. The rat skin fragment was positioned in the space between the diffusion cell compartments, with its epidermis facing the donor compartment. (18)
Mechanism of drug release:
The mechanism of drug release of the prepared transdermal patches of Pantoprazole was calculated by using the Korsmeyer equation (log cumulative percentage of drug released vs log time), and the exponent ‘n’ was calculated through the slope of the straight line. (19)
RESULT AND DISCUSSION:
Recently, there has been a lot of interest in oral site-specific drug delivery systems for the local treatment of various intestinal illnesses as well as for enhancing systemic absorption of medications, which are unstable in the stomach. However, the gastrointestinal tract's milieu and different absorption methods typically provide formulation scientists with challenges when developing and optimizing oral drug administration. Different mixes and ratios of hydrophilic and hydrophobic polymers were employed in the placebo batches. However, the HPMC E5:PVP K 30 and HPMC E5:Eudragit L 100 were chosen for the subsequent formulations with 1:1, 1:2, and 2:1 based on the production of a smooth, transparent, uniform, and flexible film.
REFERENCES:


Utsav Pathak, Chinu kumari, Dev Prakash Dahiya, A Research Study On The Formulation And Evaluation Of Pantoprazole Sodium Transdermal Patches For The Treatment Of Peptic Ulcer, Int. J. of Pharm. Sci., 2024, Vol 2, Issue 6, 919-924. https://doi.org/10.5281/zenodo.11774336
 10.5281/zenodo.11774336
10.5281/zenodo.11774336
