
We use cookies to ensure our website works properly and to personalise your experience. Cookies policy
Pulla Institute of Pharmacy, Domadugu, Sangareddy, Telagana 502313.
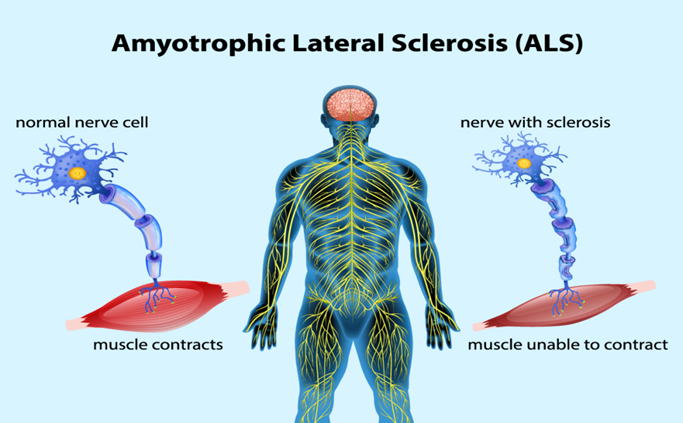
The neurodegenerative disease known as amyotrophic lateral sclerosis (ALS) typically affects the motor system, while extra-motor symptoms are becoming more widely acknowledged. Progressive muscle weakness and wasting are caused by the death of upper and lower motor neurons in the motor cortex, brain stem nuclei, and the anterior horn of the spinal cord. ALS frequently begins in a specific area and then spreads to other parts of the body, where respiratory muscle failure usually limits survival to two to five years after the disease first manifests. Extra-motor symptoms, including behavioral abnormalities, executive dysfunction, and linguistic issues, can occur in as many as 50% of cases. These issues are severe enough to meet the clinical criteria for frontotemporal dementia (FTD) in 10% to 15% of people. An autosomal dominant inheritance pattern is suggested by the family history of 10% of ALS patients. The remaining 90% are categorized as having sporadic ALS and do not have any family members who are affected. Only a portion of the causes of ALS are known, and they seem to be diverse. More than 20 genes have been linked to ALS thus far. A hexanucleotide repeat expansion in the C9orf72 gene is the most frequent genetic cause of ALS, accounting for 30% to 50% of family cases and 7% of sporadic cases. These expansions highlight the molecular similarities between ALS and FTD and are also a common cause of frontotemporal dementia. There is currently no cure or effective treatment for ALS, and multidisciplinary care—which includes symptom management, respiratory and nutritional support, and more—remains the cornerstone of treatment. Numerous facets of ALS are covered in this study, including its epidemiology, aetiology, pathophysiology, clinical characteristics, differential diagnosis, investigations, treatment, and prospects for the future.
The progressive, lethal neuromuscular illness known as amyotrophic lateral sclerosis (ALS) is characterized by degeneration of the upper and lower motor neurons that cause the body's somatic muscles to malfunction. The French neurologist Jean-Martin Charcot first used the phrase "amyotrophic lateral sclerosis" in the 1800s. [1] In the past, ALS has been divided into two categories: familial and sporadic. Due to the discovery that over 30 distinct genes are connected to the familial form of ALS, the disease has been reclassified as a multidomain neurodegenerative syndrome of motor and extra-motor systems with a variety of underlying pathophysiological mechanisms and distinct clinical sub phenotypes. [2] Increasing our knowledge of ALS's pathophysiology appears to be essential for extending life expectancy, since this will enable the creation of early and targeted diagnostic techniques. It is imperative to develop treatments that address the secondary effects of starvation and respiratory failure in addition to slowing the course of the disease. [3] Approximately 50% of ALS patients experience mild cognitive and/or behavioral disorders, and 5–15% develop dementia, typically frontotemporal. About 90% of ALS cases are sporadic of unknown origin, with the remaining 10% being classified as familial. Additionally, a variable degree of spasticity and pseudobulbar affect can be observed.[4] The failure of corticospinal (upper) motor neurons causes spasticity and rigidity in the muscles. Muscle twitches (fasciculations) are caused by the initial increased electrical excitability of afflicted lower motor neurons. As these neurons degenerate, they lose synaptic contact with their target muscles, which causes the muscles to atrophy.[5] The illness is frequently known as Lou Gehrig's disease in the US, after the New York Yankees baseball star, player who received an ALS diagnosis at the Mayo Clinic in 1939. Other neuroanatomical areas, such as the frontal cortex, may potentially be impacted in addition to motor neurons. [6] Non-invasive ventilation (NIV) has been an essential part of the treatment of ALS since 2006. NIV significantly improves cognitive function, life quality, and survival. [7] Between one and two new instances per 100,000 persons every year in the US and Europe, according to reports. Neuromuscular weakness often develops in the middle to late 50s and affects more men than women. [8] In spite of being rather A tiny proportion of ALS cases are familial in origin and linked to gene mutations, but the rest are sporadic and have a multifactorial aetiology. The pathogenesis of ALS has been better understood thanks to recent studies, which have also raised questions about whether the disease is linked to axonopathy, proteinopathy, ribonucleopathy, the neuronal microenvironment, or another disorder. [9] Currently, edaravone and riluzole are the two approved medications for the condition. Riluzole boosts survival by no more than two to three months and has no effect on quality of life. Edaravone, on the other hand, somewhat improves patient movement, however it's unclear how this impacts survival. The following seven genes have been connected to ALS since 2014: MTR3, CHCHD10, TBK1, TUBA4A, NEK1, C21orf2, and CCNF. The speedy identification of several novel genes associated with ALS is an indication of the progress in sequencing technology and, more importantly, presents an opportunity to gain a better understanding of the disease. [10]
CLASSIFICATION:
There have been several attempts to categorize motor neuron diseases according to their pathologic traits, clinical characteristics, age at start, and causation, but none of them are entirely satisfactory. The inability to identify a classification that is generally accepted as adequate or suitable highlights the existing lack of comprehensive understanding of the pathophysiologic features of each condition.[11] When a patient exhibits the clinical signs of ARC loss, the doctor must first differentiate between primary or idiopathic ARC degeneration and loss, disorders that mimic ARC loss, and those in which ARC loss is caused by the underlying pathologic condition. The potential beneficial laboratory investigations; the usage of certain tests varies depending on the patient's particular symptoms. Causes of symptomatic motor neuron disease should be investigated if the patient does have one. When there is no known cause, disorders that may have additional pathologic characteristics in addition to ARC loss must be taken into account. Once these options have been ruled out, the remaining conditions are either primary or idiopathic disorders of the ARC, which are primarily categorized based on clinical symptoms and age of start. This review focuses on the familial and sporadic variants of MND/ALS. Since there are no pathognomonic clinical symptoms or specific diagnostic tests available, the diagnosis of MND/ALS is one of exclusion. Despite this, false-positive results seldom confuse the clinical and electrophysiologic diagnosis of MND/ALS, and doctors rarely differ about the diagnosis in particular instances.[12]
EPIDEMIOLOGY
For the most part, MND/ALS can be categorized into the traditional sporadic and familial variants as well as the Western Pacific (Japan's Marianas and Kii peninsula)[13]. The Western Pacific form's clinical, pathologic,[14] and epidemiologic characteristics[15] differ from the traditional sporadic and familial variants in enough ways to warrant consideration as distinct illnesses. There have been reports of additional high-incidence foci of disorders similar to MND. However, the endemic encephalomyelitis identified in the Vilyuisk region of Siberia[16] differs from the Western Pacific and classic variants in a number of clinical and neuropathologic aspects. It is challenging to determine where West New Guinea[17] and Groote Eylandt (Australia)[18] should be included in the AHC's nosology of disorders because to a relative lack of data. Forms of the Western Pacific are found in geographically isolated areas with significantly higher occurrence.[14,15] Even greater rates of dementia and Parkinson's disease are linked to the high frequency of ALS.[19]and the prevalence of both illnesses may be rapidly shifting. In recent decades, the frequency of Parkinson's disease-related dementia appears to have been steady, whereas the incidence of ALS is said to have decreased significantly in the Marianas.[20,21] Alterations in the age at onset and the clinical signs of the "complex" of ALS, Parkinson's disease, and dementia point to a significant environmental influence and a cohort effect.Affected individuals' lifelong affinity for traditional Guamanian cuisine has raised the possibility that the neurotoxic cycad nut plays a significant role in the onset of the illness, however this theory has been challenged.[22]
The typical form of MND/ALS, on the other hand, is found all over the world and, up until recently, exhibits only minor fluctuations in incidence rates. [23] Possible causes of some of the observed differences include variations in case ascertainment. The rate of false-negative diagnostic mistakes is hard to calculate precisely,[24] even while false-positive diagnostic errors in MND/ALS are thought to be uncommon, especially at specialist referral centers. Kondo and Tsubaki investigated the frequency [25]and it can be deduced from Brownell and associates'[26] postmortem neuropathologic examinations to be at least 10% of Mayo Clin Proc, January 1991, Vol 66. Studies with older people are likely to experience false-negative mistakes the most frequently.[24,27] The highest rates (more than 2 per 100,000 people annually, adjusted for age and sex) are reported in areas recognized to have superior health-care resources. The incidence and mortality rates for MND/ALS are based on thorough case ascertainment.[28,29] Furthermore, follow-up research conducted in Minnesota's Olmsted County indicates that age-specific risk rises as people age.[29] The age-specific risk of acquiring MND/ALS appears to reduce in later age groups, and reported incidence and mortality rates are much lower than 2 per 100,000 population annually (adjusted for age and sex) in regions where case ascertainment is expected to be less comprehensive.[30,31] These findings provide credence to the theory that one significant factor influencing age-specific MND/ALS incidence and mortality rates is the thoroughness of case ascertainment.[25,32] There is also further corroborating evidence from longitudinal epidemiologic research that demonstrate that the death rate has risen in Israel[33] and most other countries in recent decades. The older population has seen the fastest growth in age-specific rates of MND/ALS in the United States and France.[23,34] Males are more likely than females to have MND/ALS, according to all published studies. However, Kahana and Zilber[33] discovered that in Israel, the sex ratio has decreased near 1:1 due to an increasing awareness of MND/ALS among elderly women, while Kondo and Tsubaki,[25] identified more false-positive diagnoses among males than among females. Although the disease can cluster geographically, conjugally, and familiarly outside of the Western Pacific region, only familial clustering is currently thought to have etiologic (most likely genetic) importance (see the discussion that follows). Numerous case-control studies have looked at possible risk factors for MND/ALS development. Although several statistically significant risk factors have been identified, only a small number have been validated by further research. Both prospective and retrospective investigations have found that antecedent trauma is a risk factor. Evidence from the latter trial, however, indicated that patients remembered past trauma more meticulously than control subjects.[35,36]
CLINICAL FEATURES
Symptomatic weakness is the first complaint in the majority of ALS patients.[37] S Limb weakness has been more prevalent than bulbar muscle weakness in every documented series.[38] Perhaps the cervical region's ARCs are more susceptible because upper limb muscles[39] are first used more often than lower limb muscles. Weakness, atrophy, and other abnormal neurologic symptoms follow an asymmetric pattern.[40]vary from one to patient and is frequently focused.[41] Although muscle cramps, paresthesias, and even pain are common ALS complaints, they are rarely the reason a patient first seeks medical attention.
While it is hard to measure, ALS progression is nearly inevitable. The significant range in illness duration suggests that there are vast variations in the rate of ARC degeneration among patients.[37] Reports of progression halting or even reversing have been made in certain patients. [42] ALS patients' respiratory dysfunction increases exponentially, at least at the end of the disease's progression,[43] according to serial measures of pulmonary function. Using mostly isometric strength assessments and a composite scoring method,[44] Munsat and colleagues recently studied 50 ALS patients[41] and discovered a linear increase of weakness in ALS during the illness's symptomatic phase. Although they acknowledged that there was significant diversity in the pace of progression among individuals, they discovered that this variance had nothing to do with the age or location of onset in any given patient. There have been prior reports linking both of these factors to a worse prognosis and, implicitly, a quicker course of the illness. [45–47]. Andres et al. discovered that bulbar function declined at the slowest pace, while respiratory and arm muscle performance declined at the highest rate.[44] Increased CSF fluid protein concentration (albeit this discovery contradicts a prior publication)[48] and severe hyperreflexia are additional clinical variables linked to a poor prognosis in Motor Neuron Disease. [46] Despite the fact that MNDIALS is primarily a motor system degeneration, researchers have looked for evidence linking seemingly unrelated parts of the nervous system in their quest for causative clues.[49] Although there have been reports of oculomotor neuron loss from the brain stem and a histologically confirmed case of ocular palsy in an ALS patient,[50] the extraocular muscles are almost never implicated in ALS symptoms. When life-support techniques are used to prolong a patient's life, oculomotor neuronal abnormalities may become more common."[51] A significant number of MND/ALS patients have anomalies in saccadic velocities,[52] ocular smooth pursuit, and visual scanning that can be identified both clinically and instrumentally. However, it's unclear what the pathogenic basis of these aberrations is. Although it is uncommon, reports of clinically severe nystagmus have been made.[53] Depending on the criteria used to characterize the phenomena, up to 25% of individuals may experience sensory symptoms at some point throughout their disease, which are commonly reported by patients with MND/ALS[49,54-56]. However, objective clinical sensory indicators are rare and raise questions about the diagnosis's precision when they do occur.[54] When quantitative techniques and computer-assisted sensory examination are used, the frequency of objective sensory abnormalities rises. According to the latter approach, identifiable anomalies are seen in over 15% of MNDIALS patients.[57]
Even in the later stages of the disease, control over the bladder, intestine, and autonomic nervous system is thought to be unaffected. Just 3.2% of patients in a population research experienced loss of sphincter control unrelated to prostatic enlargement,[37] which is comparable to the 4% of patients in a recent clinical series who experienced otherwise unexplained incontinence. [58] However, many people have been discovered to have lesser degrees of sphincter dysfunction when asked direct, careful questions. [59] The significance of the described symptoms is unknown because it is thought that MND/ALS[60] preserves the ARCs that regulate the external sphincters. Two recent investigations discovered subclinical abnormalities in autonomic function in a large number of individuals using a variety of quantitative measurements.58,61] According to one of these reports, autonomic dysfunction was linked to faster progression,[58,59] and 38% of patients reported aberrant results on the Valsalva maneuver or the quantitative sudomotor axon reflex test. The idea of an etiological relationship between the Western Pacific type of ALS and the sporadic and familial variants prevalent in other geographic locations is raised by the correlation between ALS, dementia, and Parkinson's disease.[62,63] Only slightly more than 6% of ALS patients received a diagnosis in a population-based research conducted in Israel,[37] and the incidence of dementia in ALS patients has been found to be low[26,49] Compared to control subjects, neither Parkinson's disease nor dementia were more frequently included on the death certificates of patients with MND/ALS.[64]
ETIOLOGY
In about 90 to 95 percent of cases, ALS occurs sporadically (7 sALS). The remaining patients, approximately 5–10%, have a positive family history and are diagnosed with familial ALS (7 fALS)[65]. Here we describe autosomal-dominant and autosomal-regressive erbgänge. About 10–15% of fALS patients have a mutation on chromosome 21q22 in the gene for the enzyme zytosolischen Kupfer-Zink-Superoxide-Dismutase (Cu-Zn-SOD), which causes free radicals to be released (fALS1). Over 100 mutations, most of which are autosomal-dominant, have been described to date. These can only be considered a primary cause of the disease in about 1% of ALS patients overall, but when combined with the appropriate transgenic Tier-Model, transgenic mice, and mutations in the human Cu/Zn-SOD gene, they provide a crucial foundation for the acquisition of new knowledge about the pathophysiology and biochemistry of the disease[66]. Here, it appears that the degeneration of motor neurons is caused by a toxic function of the mutated Cu/Zn-SOD-Gens. Seven mitochondrial leaks and resulting disruptions in energy-strategy (possibly caused by or a result of increased exposure to free radicals) as well as heightened cellular susceptibility to excitatory neurotransmitters (glutamat) were observed in the Tier-Model. The relevance of these model representations lies in the applicability of therapeutic strategies such as antioxidant use or antiexzitotoxic principles. Ben Hamida was the first to describe fALS2, a juvenile, autosomal-reversible form of ALS [67]. She starts i.d.R. before the age of 20, progresses gradually, and enters v.a. with the signs of a 1. Motoneurons. Kopplungs analyses have demonstrated that the responsible gene is located on chromosomes 2q33–q35 and is coded by a GTPase called Alsin. A rare autosomal-dominant vererbte juvenile, slowly progressive ALS form was first described in 1998 as a result of a mutation in the SETX (Sentaxin) genes on chromosome 9q34 (fALS4) [66]. The autosomal-dominant vererbte ALS known as fALS8 is characterized by a delayed onset, slower progression, and an earlier loss of two motor neurons. It is caused by a mutation on chromosome 20 in the gene for vesicle associated membrane protein B (VAPB), a protein that is crucial for intracellular membrane transport [66,68].Furthermore, it was demonstrated that seven mutations in the Angiogenin-Gen (Angiogenin is a protein that contributes to the development of blood vessels) correlate with the onset of amyotrophen lateral sclerosis [69]. The significance of two other genes for the pathogenesis of ALS is still unknown: Possible mutations in the 7-Dynactin gene (encoded for a protein associated with axonal transport) have been linked to the development of amyotrophen lateral sclerosis [70], and polymorphisms in the 7-VEGF ("vascular endothelial growth factor") gene promoter have also been identified.[71]
PATHOLOGIC FEATURES
The central nervous system Recent research has examined macroscopic, microscopic, and ultrastructural aspects of the pathologic alterations in MNDIALS.[ 76,72,73] Although atrophy of the motor and premotor cortices is typically evident, the brain may appear normal macroscopically in ALS. The most noticeable alterations include palpable stiffness (gliosis) of the lateral columns, which is how ALS is termed, and atrophy of the spinal cord and related ventral roots, particularly at the level of the cervical and lumbar expansions. These characteristics are often most noticeable in the caudal areas, and the loss of ventral root fibers is typically accompanied by muscle atrophy.[72] Degeneration and loss of AHC neurons is the main characteristic of ALS. This process's pathogenic characteristics include neuronophagia, gliosis, shrinkage, and pyknosis (which causes lipofuscin to become more prominent).?[72] "Because of a massive loss of Betz cells and other pyramidal cells from the precentral cortex, and to a lesser extent from the postcentral cortex, the remaining feature is appreciable reactive gliosis."[74] The loss of cortical cells is accompanied by a selective depletion of big myelinated axons in the corticospinal tracts.[75]
The anterior and lateral spinal cord columns, especially caudally, are where cortico-spinal tract involvement is most easily observed. This involvement varies and is rarely absent (although this varies according on how one defines ALS and MND). In the brain stem and cerebrum, degeneration is present in these same long-tract fibers more rostrally, however it is technically challenging to show. The sporadic type of the disease typically (though not always) spares the posterior columns.[14,37] It can be challenging to assess cell loss in the brain-stem nuclei, and related observations such intracellular degenerative alterations and target muscle denervation would be required to validate pathogenic involvement in this area. Compared to the upper brain-stem nuclei, the lower brain-stem nuclei are nearly always more widely and persistently involved.[72] The oculomotor nuclei typically exhibit pathologic intracellular alterations, but nerve fiber loss is mild and infrequently accompanied by clinical symptoms.[77] The spinal cords anterior horn exhibits the most noticeable neuronal alterations. Atrophy of the big spinal motor neurons, followed by degeneration, loss, and glial replacement, are the main alterations.[78] The remaining cells could be mostly normal, slightly or significantly atrophied, or include a range of inclusion bodies (explained in more detail below).[73,78]On rare occasions, one may notice vacuolation, characteristic central chromatolysis, or neuronophagia. Previously, it was claimed that Clarke's nucleus' neurons were unaffected. But according to quantitative techniques, Clarke's nucleus is frequently affected, with a mean 36% loss of neurons and neurofilamentous degeneration in some of the cells that survive. These neuronal alterations are accompanied by degeneration of the spinocerebellar tracts.[79] The motor neurons in Onufrowicz's sacral nucleus, which are thought to innervate the anal and urethral external sphincters, have been carefully studied and found to be universally retained in MND/ALS.[48,80] It has also been proposed that this nucleus supports certain autonomic processes. [80] Late involvement of the oculomotor and sacral nuclei has been observed in individuals whose course of disease has been prolonged by mechanical ventilation and parenteral nutrition, in addition to the understanding that the neurons of Clarke's nucleus degenerate in MND/ALS.[39] According to this research, these neuronal phenotypes are more resistant to the pathophysiologic process than lower cranial and spinal motor neurons, but they are nevertheless susceptible to it.
CYTOLASMIC AND ULTRASTRUCTURAL FEATURES
In MND/ALS, several cytoplasmic and ultrastructural anomalies have been found. None of them are pathognomonic, despite the fact that some are typical of the illness and others are frequently present in it. [73] Spheroids are irregular swellings of material associated with ARCs that have a whorled fibrillary pattern and are markedly argentophilic (silver impregnation stain) and mildly eosinophilic (hematoxylin-eosin stain).[81,82,83]However, because they are present in normal spinal cords, rats with "W-iminodipropionitrile" or aluminum toxicity, and in hereditary canine spinal muscular atrophy, they are not unique to the illness.?[84] The anterior horns of the control and ALS spinal cords can both include spheroids, but the latter's presence is noticeably more numerous.[81,85]Spheroids can develop in either dendrites or axons and have globular, spindle-shaped, or beaded appearances.[86] They appear to be most prevalent early in the course of the illness, which makes them particularly interesting.[86,87] and are linked to the atrophy of the process in which they occur when they are large. Spheroid development and neuronal degeneration may be complimentary processes, as evidenced by the observation that spheroid volume appears to rise in proportion to atrophy of the cell soma and either chromatolysis or an increase in lipofuscin prominence.
Spheroids are made up of interwoven bundles of IO-nm neurofilaments at the ultrastructural level. They contain cytoplasmic features like mitochondria, pieces of smooth endoplasmic reticulum, and, in rare cases, entire nuclei, along with other, more odd structures like Hirano bodies and polyglucosan bodies,[85] which appear to have been trapped during the spheroid's growth. According to a 1987 study[88], there were no differences between the spheroids of control and ALS cases, and the immunostaining characteristics of the phosphorylated and non-phosphorylated neurofilament proteins in globules and spheroids were identical.
Recent studies have revealed that the soma and proximal axons of ARCs from ALS patients have a comparatively higher number of phosphorylated neurofilaments detected by monoclonal antibodies; this finding implies that premature or excessive neurofilament phosphorylation may be linked to impairment of neurofilament transport.[76,73] Bunina bodies are small (2- to 3-Ilm), round, slightly refractile eosinophilic structures found in the neuronal cytoplasm of a significant percentage of patients with MND/ALS.2,77 Four to six of them are usually found in a neuron, and they can combine to form chains or filigrees. Although their exact origin is uncertain, they may be autophagic and are most likely not unique to MND/ALS.[76]Due of the tight relationship of transitional forms, Chou'' thought that Bunina bodies originated from basophilic inclusions that were thought to possess high RNA concentrations. Though their ultrastructural characteristics are diverse, basophilic inclusions have a lot of rough endoplasmic reticulum and a lot of ribosomes. In one instance of spontaneous MND/ALS with a remarkably brief course (only 10 months), chromatolysis, spheroid development, neurofilament buildup, and Bunina bodies were discovered. Since chromatolysis and spheroid formation were more noticeable in this instance than in instances that were later studied for the same characteristics, Hirano[87] suggested that these might be early pathologic alterations in MND/ALS.
PERIPHERAL NERVOUS SYSTEM
Recent research on the spinal and ranial motor nerve roots, as well as the hypoglossal and phrenic nerves,[77] has validated and extended the idea that massive myelinated fibers are lost from the ventral spinal roots[91] in ALS. P"Thoracic and sacral spinal cord outflow is relatively spared."[77] Studies on teased fibers support the significantly higher incidence of myelinated fiber degradation and the ensuing subsequent segmental de-myelination.[77,91] Furthermore, some unmyelinated fiber degradation has been seen. [91] It has been reported that the quantity of small-diameter myelinated fibers has either been conserved or even increased. [90,91]
These results are in line with a pathologic process in MND/ALS that is mostly neuronal (as opposed to axonal, or "dying back"), with gamma motor neurons largely preserved. Axonal atrophy or the existence of smaller, regenerating fibers could be the cause of an increase in small myelinated fibers. "In intra-muscular nerves, axonal atrophy may be a major additional factor." The phrenic nerve is the primary example of the latter interpretation supported by current findings. The neuropathologic involvement of peripheral sensory neurons in at least.[93] Some cases of MND/ALS appear to be unquestionable, in contrast to the clinically insignificant symptoms and indicators of sensory disruption.[42,91] Large myelinated fibers in the sura and superficial peroneal nerves deteriorate and become fewer in number, and there is evidence that unmyelinated fibers are also impacted. Lumbar dorsal root ganglion cells are destroyed.[91]
CURRENT OF ALS FOR FUTURE MEDICATION DEVELOMENT
ALS is thought to be a complicated, multifactorial, and multisystem disease based on prior research from ALS patients, transgenic animal models, and in vitro investigations. Numerous processes, including oxidative stress, glutamate excitotoxicity, mitochondrial damage, protein aggregation, glia and neuroinflammatory disease, impaired axonal transport, and altered RNA metabolism, have been linked to the pathophysiology of ALS.[94]Oxidative stress.
TREATMENT
Fingolmod : An oral sphingosine-1-phosphate-receptor modulator called tingolimod (Gilenya, Novartis, Dorval, Canada) is presently undergoing testing for the treatment of ALS. It controls migration, adhesion, differentiation, proliferation, and adhesion via acting on G-protein couple receptors. Additionally, there is proof that fingolimod works by stopping lymphocytes from leaving lymph nodes [95]. As a result, fewer potentially autoaggressive lymphocytes infiltrate the central nervous system [96]. Fingolimod has been shown to exhibit neuroprotective qualities in in vitro models of ALS [97]. To compare the acute safety and acceptability of oral administration of 0.5 mg fingolimod to the corresponding placebo, a randomized, double-blind, phase II research was recently designed. Participant recruitment for this study has begun, and it is currently underway.
Ariclomol : By increasing the expression of the heat shock protein gene, ariclomol , an analog of bimoclomol, causes heat shock protein to be produced during cell stress. In an animal model of ALS, arimoclomol has been demonstrated to prolong life [98].200 mg of arimoclomol for FALS patients who have SOD1-positive results. The major and secondary outcome measures that were designed were the rate of decrease of the ALS Functional Rating Scale-Revised (ALSFRS-R) over a 12-month period, the rate of reduction in forced expiratory volume in 6 seconds as a measure of disease progression, and the estimation of motor unit numbers.
Rasagiline : Rasagiline, an MAO-B inhibitor used to treat Parkinson's disease, is manufactured by Teva Pharmaceutical Industries Ltd. in Petah Tikva, Israel [99]. Animal survival and preclinical motor abilities were significantly impacted by the medication in a dose-dependent manner [100]. To examine the effectiveness of rasagiline as an adjuvant medication to riluzole in the treatment of ALS, a phase II multicenter, randomized, double-blind experiment is now underway. The trial's main objective is to assess survival time; quality of life and ALSFRS change are secondary end measures. (t is still in process)
Ozanzumab : A monoclonal antibody called ozanezumab works by blocking neurite outgrowth inhibitor-A (NOGO-A). Preclinical research showed that neuro-muscular junction integrity is impacted by high NOGO-A expression, and that destabilizing and suppressing nerve terminals improved the health of motor neurons in mice, suggesting that NOGO-A plays an important role in ALS [101]. The effectiveness and safety of intravenous administration of ozanezumab in comparison to a matching placebo were examined in a phase II, multicenter, randomized, double-blind study. Clinicaltrials.gov: Study of Ozanezumab (GSK1223249) vs. Placebo in the Treatment of Amyotrophic Lateral Sclerosis, https://clinicaltrials.gov/ct2/results?term=NCT 01753076&Search= Search evaluates the impact of intervention on the function and survival duration of ALS patients.
Stem cell therapy in ALS :
Recent advances in stem cell research, however, may open the door to neural implantation and cell replacement therapy for ALS patients. The promise of cell implantation or transplantation is that it may help compensate for the central nervous system's incapacity to regenerate destroyed neurons. Several in vivo models of ALS have shown promising outcomes from stem cell research employing diverse stem cell types. There is the strongest evidence supporting the use of neural progenitor cells (derived from the spinal cord) and mesenchymal stem cells (derived from bone marrow) in ALS. In an animal model with SOD1, both neural progenitor cells and mesenchymal stem cells performed well and demonstrated increased survival in comparison to control mice.[102]Only a tiny number of human investigations have yielded positive findings; the majority of this research used small, nonrandomized samples [103]. Before stem cell therapy for ALS can ever be considered as even an experimental therapeutic method, much more work needs to be done. In September 2013, Drs. Eggan and Rubin, two scientists from the Harvard Stem Cell Institute, joined forces with Evotec, a German biotech company, to screen potential drugs in their ALS stem cell models. The goal was to swiftly identify molecules that could be developed into ALS treatments.
Human neural stem cells :
It is possible for human neural stem cells (HNSCs) to constantly proliferate and differentiate into various neuronal, astrocyte, and oligodendrocyte types. ALS, Parkinson's disease, Alzheimer's disease, and traumatic brain damage such cerebral stroke are among the neurodegenerative illnesses for which HNSCs are being extensively investigated. According to [104], HNSCs have the capacity to migrate and replace the dying neurons in the central nervous system. By delaying muscle degeneration, HNSCs demonstrated encouraging outcomes in a number of preclinical investigations [105]To test the safety of the intervention, a phase I trial was carried out in which 18 ALS patients received a dose of 1/10 of the cell dose that is meant to be used for therapeutic purposes. Phase I study results showed that the intervention was safe to use and that quality of life had greatly improved. Phase II dose escalation and safety investigations were authorized by the FDA to begin.
Ventilation therapy :
Breathing becomes difficult for most ALS patients because their respiratory muscles deteriorate. Patients with ALS struggle to draw oxygen in and expel carbon dioxide into the atmosphere, in contrast to those with other respiratory conditions. As a result, the blood's oxygen content decreases and its carbon dioxide content increases. As a result, using more oxygen is typically inappropriate. The use of either intrusive ventilation, such as a ventilator, or noninvasive mechanical ventilation, such as intermittent positive pressure ventilation or bilevel positive air pressure, is the solution to this issue in ALS patients. ALS patients' quality of life and survival were enhanced by the use of mechanical ventilators [106].
Psychotherapy :
ALS patients frequently experience depression [108,107]; the majority of patients experience depression upon the diagnosis of the illness. Antidepressant therapy and patient counseling will help patients and their loved ones manage. In addition to prescribing antidepressants, psychological treatment may be required at times; this is typically best administered by family members. It is advisable to use drugs like fluoxetine that have little anticholinergic action. One of the tricyclic antidepressants will be more beneficial if there is persistent pain, excessive sweating, excessive salivation, or sleep difficulties [108].
CONCLUSION :
ALS is still a neuromuscular illness that kills people. The ideal biological pathways to use as novel targets for therapeutic modulation of motor neuronal death in ALS will only be determined in the future. One of the biggest obstacles to neurologists providing timely, appropriate treatment for ALS is early and correct diagnosis. Riluzole is still the sole pharmacological treatment for ALS that has been shown to be successful and that NICE recommends, despite intensive study, and that may stop the disease's development. Therefore, there is an urgent need for novel therapies that address the underlying illness. There are currently a number of encouraging randomized clinical studies in progress, and greater cooperation between pharmaceutical companies, basic and clinical researchers, neurologists, and psychologists may advance the development of the best possible treatment for ALS. Using a single drug that targets many disease pathways or combining drugs with various modes of action would likely result in the highest therapeutic results. The exact substance that will be the most successful ALS treatment .
REFERENCES




Padala Ramesh, Aleena Maria Martin, Maddula Nikitha, Gavindla Manisha, A Review Article on “Amyotrophic Lateral Sclerosis”, Int. J. of Pharm. Sci., 2025, Vol 3, Issue 10, 293-309. https://doi.org/10.5281/zenodo.17255492
 10.5281/zenodo.17255492
10.5281/zenodo.17255492