
We use cookies to ensure our website works properly and to personalise your experience. Cookies policy
P.R. Patil Institute of Pharmacy, Talegaon (S.P), Ashti – 442202, Wardha, Maharashtra, India.
Analytical method validation is an essential process in pharmaceutical analysis, ensuring that analytical methods are suitable for their intended purpose and comply with International Council for Harmonization (ICH) guidelines. A validation study is designed to provide sufficient evidence that the analytical procedure meets its objectives. These objectives are described with a suitable set of performance characteristics and related performance criteria, which can vary depending on the intended purpose of the analytical procedure and the specific technology selected [8]. The validation process provides documented evidence of accuracy, precision, specificity, linearity, range, detection limit, quantitation limit, robustness, and system suitability. These parameters collectively ensure the reliability and reproducibility of analytical data during drug development, quality control, and regulatory submissions. This review discusses the principles, requirements, and methodology of analytical method validation as outlined in ICH Q2(R1) and the revised Q2(R2) guidelines. Emphasis is placed on the importance of protocol-driven validation and its role in achieving consistent pharmaceutical quality. The discussion also highlights best practices for designing and executing validation protocols, covering both small molecule and biotechnological products [1,2,3].
The primary aim of any pharmaceutical manufacturing unit is to consistently produce medicines of the required quality at the most economical cost [9]. The role of Validation is crucial, as it ensures reliable and repeatable outcomes for both routine analysis and stability testing. This has become increasingly important in the field of quality control and accreditation, particularly with the growing focus on dissolution, testing and impurity profiling in recent years [10]. The concept of validation was first introduced in the United States in 1978.
VALIDATION:
As defined by the U.S. Food and Drug Administration (FDA), validation refers to a process of production and process control intended to confirm that drug products consistently maintain their identity, strength, quality, and purity. According to the FDA guidelines issued in May 1987, a validation dossier must contain all the necessary data and testing procedures that demonstrate the system and process comply with established requirements [11]. The word validation means assessment of validity or action of proving effectiveness [12].
Analytical methods need to be validated or revalidated.
i.Before their introduction into routine use.
ii. When there is a change in the conditions under which the method was originally validated (for example, use of an instrument with different specifications or samples having a different matrix).
iii. When any changes are made to the method that falls beyond the scope of the initial validation [13].
Significance of Validation:
Parameters of Analytical Method Validation [22]
The primary purpose of method validation is to demonstrate that an analytical method performs as intended, giving results that are accurate, reliable, and consistent. Validation of analytical methods is carried out in line with the International Council for Harmonization (ICH) guidelines, specifically ICH Q2.
The validation parameters are
• Accuracy
• Precision
• Specificity /Selectivity
• Limit of Detection
• Limit of Quantitation
• Linearity
• Range
• Robustness
• Ruggedness
Accuracy
Accuracy of an analytical method refers to how close the results obtained are to the true or accepted reference value. It indicates the degree of correctness of the analytical procedure. In simpler terms, it measures how close the experimental value is to the actual value. Accuracy is usually expressed as the percentage recovery of a known quantity of analyte within the specified linearity range. [9]
Accuracy of an analytical method is evaluated at different concentration levels (80%, 100%, and 120%) to ensure reliability across the range.
It can be confirmed by comparison with a validated method and should typically be within 3–5% of the true value. As per ICH guidelines, accuracy is assessed using nine determinations at three concentration levels. [17]
The Recovery is determined by the equation:
Recovery = Analytical Result/True Value * 100%
Limit: The recovery should be in the range of 98.0% to 102.0%
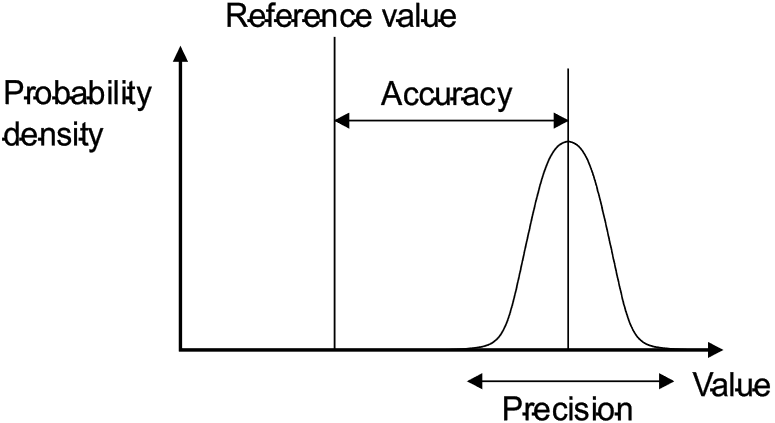
Figure 1
Precision
Precision of an analytical method refers to the closeness of agreement among a series of measurements obtained from multiple analyses of the same homogeneous sample under defined conditions. It indicates the degree of repeatability or consistency of the results when the procedure is repeated. [16]
% RSD =Standard Deviation/Mean x 100
Where, RSD = Relative Standard Deviation
Acceptance Criteria
At each concentration level, the precision of the analytical method should have a coefficient of variation (CV) not greater than 15%. However, for the lower limit of quantification (LLOQ), the CV may be up to 20%, reflecting acceptable variability at low concentration levels. [15]
Specificity / Specificity
Specificity refers to the ability of an analytical method to accurately identify and measure the analyte in the presence of other substances that may be present, such as impurities, excipients, or degradation products. Ensuring specificity is essential during method development and validation, as a lack of it can negatively impact accuracy, precision, and linearity. It should be regularly verified throughout validation and routine application of the method. [18] Selectivity of an analytical method is defined as its ability to accurately measure the analyte in the presence of other components that may be expected to be present in the sample matrix. It represents the method’s capacity to resolve and distinguish between different chemical entities. Selectivity is typically assessed by comparing the analytical response of the analyte in the absence and presence of potentially interfering substances, ensuring reliable identification and quantification. [9]
Limit of Detection (LOD)
The Limit of Detection (LOD) is defined as the lowest concentration of an analyte in a sample that can be reliably detected, though not necessarily quantified, under specified experimental conditions. In methods using UV detectors, detecting low-level compounds can be challenging due to gradual changes in detector lamp sensitivity or variations in baseline noise. It is important to ensure that the LOD and LOQ can be consistently achieved with the method. Without a reference standard for an impurity, detection of small peaks may be unreliable. LOD can be estimated using different approaches depending on the type of method:
Visual evaluation – observing the presence of the analyte peak.
Signal-to-noise ratio – comparing the analyte signal to baseline noise.
Standard deviation of the response – using statistical calculations from multiple measurements.[19]
It can be calculated using the formula:
LOD = 3.3 s/S
where:
s = standard deviation of the response
S = slope of the calibration curve of the analyte
Limit of Quantitation (LOQ)
The Limit of Quantitation (LOQ) of an analytical method is the lowest concentration of an analyte in a sample that can be determined quantitatively with acceptable precision and accuracy.[18]
It can be calculated using the formula:
LOQ=10*s/S
where:
s = standard deviation of the response
S = slope of the calibration curve of the analyte
Linearity
Linearity of an analytical method refers to its ability to elicit test results that are directly proportional to the concentration of analyte within a given range. It is typically evaluated by analysing a series of standard solutions at different concentrations and plotting the corresponding responses to construct a calibration curve. The linearity is then assessed through linear regression analysis, where the slope, intercept, and correlation coefficient of the calibration curve provide quantitative measures of the method’s linear response. [19]
Range
The range of a method refers to the time between the highest and lowest concentrations of the analyte for which the procedure has been demonstrated to provide acceptable levels of accuracy, precision, and linearity. The range is normally expressed in the same units as the test results (e.g. percentage, parts per million) obtained by the analytical method. [17]
Minimum specified ranges:
For the assay of drug substance or finished product: 80–120% of test concentration
Content uniformity: 70–130% (or wider if justified)
Dissolution testing: ±20% of the specified range [20]
Robustness
Robustness refers to the ability of an analytical method to remain unaffected by small, intentional variations in method parameters. In the context of HPLC, these variable parameters can include factors such as the flow rate, column temperature, sample temperature, pH, and composition of the mobile phase. [14]
Examples of typical variations are:
In the case of liquid chromatography, examples of typical variations are:
In the case of gas-chromatography, examples of typical variations are:
Ruggedness
Ruggedness refers to the ability of an analytical method to produce consistent and reproducible results when subjected to variations that are commonly encountered in different laboratories or by different analysts. It assesses how reliably the method performs under changes such as different operators, instruments, reagents, laboratories, temperatures, or timing. To evaluate ruggedness, the reproducibility of results for the same sample is measured under these varying conditions and compared with the method’s precision under standard conditions. This comparison provides an indication of how robust and dependable the analytical method is in practical use. [17]
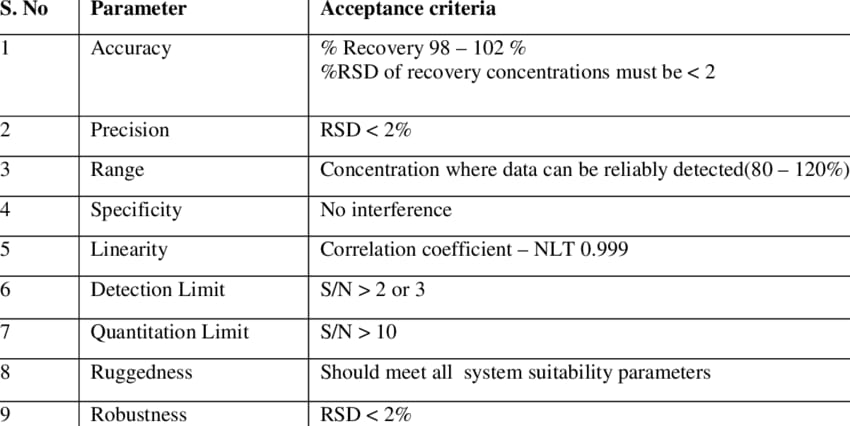
Acceptance Criteria of Validation for HPLC:
Table 1
Validation Protocol [7,8]
Validation protocol should include the following points:
CONCLUSION
The article emphasizes that analytical method validation is a crucial aspect of the pharmaceutical industry, ensuring that analytical methods are accurate, reliable, and suitable for their intended purpose. It provides a comprehensive understanding of validation, its types, necessity, development procedures, and key parameters such as linearity, LOD, LOQ, range, specificity, robustness, ruggedness, and system suitability. The review aims to guide young researchers in enhancing the quality of analytical method development and validation, thereby ensuring consistency, reliability, and overall quality in drug development and production.
Conflict Of Interest
There is no conflict of interest from all the authors.
ACKNOWLEDGEMENT
We are thankful to the management of P.R. Patil Institute of Pharmacy, Talegaon (S.P.), Wardha for providing all the facilities for carrying out this review work.
REFERENCES





P. D. Ikhar*, P. G. Gawande, P. D. Thakre, P. D. Darange, V. D. Dapurkar, A Review on Analytical Method Validation as Per ICH Guidelines and Protocols, Int. J. of Pharm. Sci., 2025, Vol 3, Issue 11, 2595-2600 https://doi.org/10.5281/zenodo.17638632
 10.5281/zenodo.17638632
10.5281/zenodo.17638632