1B. Pharmacy VIII- Semester Students, Aryabhatta Knowledge University, A and E College of Pharmacy, Baluahi, Mohiuddin Nagar, Samastipur, Bihar.
2Assistant Professor, Faculty of Pharmaceutical Sciences, A and E College of Pharmacy, Baluahi, Mohiuddin Nagar, Samastipur, Bihar.
Apoptosis is a natural, regulated process of cell death that maintains tissue health by removing damaged or dangerous cells. It is crucial in both normal body functions and disease conditions. When apoptosis is disrupted, it can lead to diseases—excessive apoptosis is linked to degenerative disorders, while insufficient apoptosis is a major factor in cancer. In cancer, the imbalance between rapid cell growth and reduced cell death allows abnormal cells to survive and spread. Interestingly, some cancers show high levels of apoptosis, which can help suppress tumor growth. However, this process is complex. In some cases, limited apoptosis can influence the tumor microenvironment (TME) in ways that actually support cancer growth and resistance to treatment. The dual nature of apoptosis—both suppressing and potentially supporting tumors—makes it a challenging yet promising target for cancer therapies. Researchers are developing drugs to enhance apoptosis, but these require thorough testing to ensure safety and effectiveness.
1.0 Introduction – Role of Apoptosis in Cancer
Apoptosis is a natural, tightly regulated process of programmed cell death vital for development and tissue maintenance. It acts as a defense mechanism by eliminating damaged or dangerous cells, and can be triggered by factors like infections, DNA damage, and cancer treatments. Central to apoptosis are caspase enzymes, activated through internal (mitochondrial) or external (receptor-mediated) pathways. In cancer, tumor cells often avoid apoptosis, helping them survive, multiply, and resist therapy. Some aggressive cancers even show high apoptosis levels, suggesting that dying tumor cells might aid tumor growth under certain conditions. The disruption of the balance between cell division and cell death is a key feature in cancer. For instance, mutations in the tumor-suppressor gene p53, which promotes apoptosis, are common. Additionally, treatments that kill cancer cells may unintentionally promote tumor recovery by altering the surrounding tumor microenvironment (TME). Therefore, effective cancer therapies must not only target tumor cells but also address their complex interactions within the TME to overcome resistance and improve treatment outcomes.[1]
2.0 Morphological Changes in Apoptosis
Apoptosis, or programmed cell death, involves distinct and orderly structural changes in both the nucleus and cytoplasm of the cell. These changes are consistent across various cell types and usually take several hours to complete, depending on the cell type and stimulus.
2.1 Nuclear Changes
2.2 Cytoplasmic and Cellular Changes
Importantly, during apoptosis, the cell’s outer membrane stays intact, preventing inflammation and distinguishing it from uncontrolled cell death processes like necrosis.
2.3 Final Stages and Clearance of Apoptosis
In the final phase of apoptosis, the cell breaks into small, membrane-enclosed fragments called apoptotic bodies, which safely contain parts of the nucleus and cytoplasm. These are quickly recognized and removed by nearby phagocytes (immune cells), ensuring no harmful substances leak out. This rapid and clean removal explains why apoptotic bodies are rarely observed under normal conditions.
2.4 Secondary Necrosis
If apoptotic cells are not cleared in time—such as in lab settings without phagocytes—they eventually undergo secondary necrosis, where the membrane breaks down. This process resembles necrosis and may lead to inflammation, unlike the controlled nature of normal apoptosis.
3.0 Biochemical Changes in Apoptosis
Apoptosis involves a series of organized biochemical events that dismantle the cell without causing inflammation. These changes are mainly grouped into three categories: caspase activation, DNA/protein breakdown, and membrane alterations aiding phagocytosis.
3.1 Activation of Caspases
Caspases are key enzymes that get activated during apoptosis. They cleave specific proteins and activate DNA-degrading enzymes, leading to controlled disassembly of the cell’s structure and function.
3.2 DNA and Protein Breakdown
Apoptotic cells show distinct DNA fragmentation. DNA is first broken into large pieces, then into smaller, 180–200 base-pair fragments forming a DNA ladder—a hallmark of apoptosis. However, this pattern is not exclusive to apoptosis and may appear in necrosis too.
3.3 Membrane Changes and Recognition by Phagocytes
A key membrane change is the flipping of phosphatidylserine (PS) from the inner to the outer membrane, signaling phagocytes to engulf the dying cell. This prevents inflammation and supports clean cell removal.[3]
3.4 Limitations and Considerations
While biochemical markers like caspase activity and DNA fragmentation help detect apoptosis, they have limitations. Not all apoptosis follows the same pathways—some may be caspase-independent. Hence, experts recommend emphasizing morphological features for accurate identification of apoptosis.
4.0 Mechanisms of Apoptosis
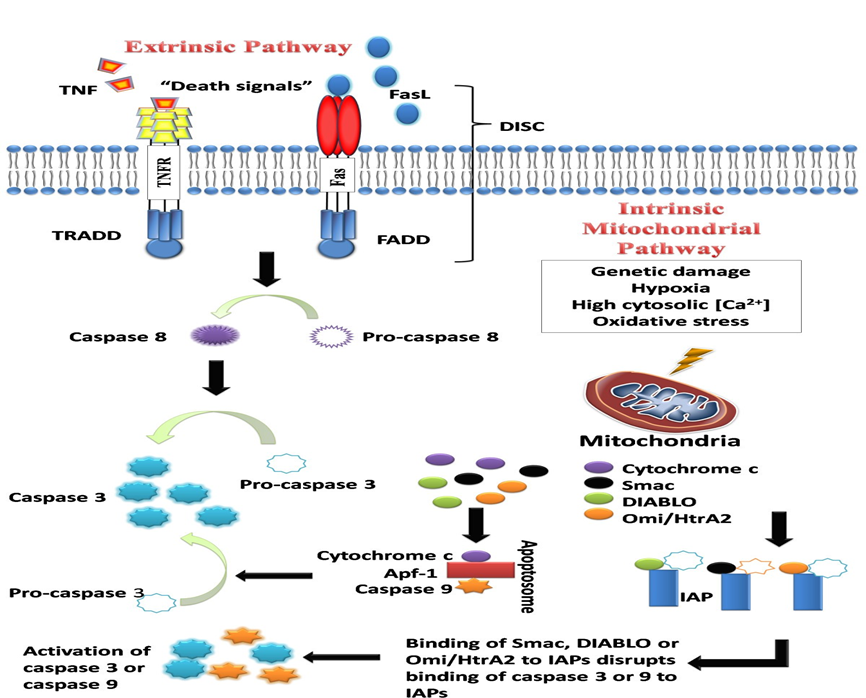
Fig 01:- The intrinsic and extrinsic pathways of apoptosis [12]
Understanding how apoptosis is triggered and regulated is crucial, as disruptions in these mechanisms are linked to many diseases, including cancer. The process is mainly driven by caspases, which act as both initiators and executors of cell death. There are three key pathways that activate apoptosis:
4.1 Extrinsic (Death Receptor) Pathway
This pathway is triggered by external signals, where death ligands (like TNF or FasL) bind to specific death receptors (TNFR1, Fas/CD95) on the cell surface. This binding forms a complex called DISC (Death-Inducing Signaling Complex), which activates caspase 8, an initiator enzyme. Caspase 8 then activates downstream executioner caspases, leading to controlled cell death.
4.2 Intrinsic (Mitochondrial) Pathway
This pathway starts from within the cell, usually due to internal stress like DNA damage, oxidative stress, or calcium imbalance. These stress signals increase mitochondrial membrane permeability, causing the release of cytochrome c and other apoptotic proteins. Cytochrome c helps form the apoptosome complex, which activates caspase 9. The pathway is tightly regulated by the Bcl-2 protein family, where pro-apoptotic members (e.g., Bax, Bak) promote cell death and anti-apoptotic members (e.g., Bcl-2, Bcl-XL) try to prevent it. Additional proteins like Smac/DIABLO and Omi/HtrA2 enhance apoptosis by blocking caspase inhibitors.[4]
4.3 The Common Execution Pathway
Both the extrinsic and intrinsic apoptotic pathways ultimately activate the common execution phase, centered around caspase 3, the key executioner enzyme. Caspase 9 (intrinsic) and caspase 8 (extrinsic) both activate caspase 3, which then cleaves the inhibitor of caspase-activated DNase (CAD), leading to DNA fragmentation—a hallmark of apoptosis. Caspase 3 also targets various other cellular components, including kinases, cytoskeletal proteins, DNA repair enzymes, and more. These cleavages cause structural and functional breakdowns in the cell, such as membrane blebbing, cytoskeleton collapse, and the formation of apoptotic bodies, ensuring an orderly dismantling process.
4.4 The Intrinsic Endoplasmic Reticulum (ER) Pathway
The ER stress-induced pathway is a third, less common apoptotic route, triggered by cellular stress like hypoxia, oxidative damage, or glucose deprivation, which leads to protein misfolding in the ER. This activates caspase 12, independent of mitochondria.[5]. Activation occurs when TRAF2 dissociates from procaspase 12, initiating a cascade that leads to apoptosis. This ER pathway provides an additional layer of regulation, allowing cells to respond to internal stress not directly involving mitochondria.
5.0 Apoptosis and Carcinogenesis
Apoptosis plays a crucial role in preventing cancer by eliminating damaged or abnormal cells. However, cancerous cells often evade apoptosis, enabling unchecked growth and survival. This evasion is considered a hallmark of cancer and occurs through several mechanisms:

Fig 02:-Mechanisms contributing to evasion of apoptosis and carcinogenesis.[13]
1.Imbalance between pro- and anti-apoptotic protein
2.Reduced caspase activity
3.Defects in death receptor signalling
These changes allow malignant cells to resist death signals, persist, and accumulate further mutations.
5.1.1 Disrupted Balance of Pro-apoptotic and Anti-apoptotic Proteins
A key regulatory group in apoptosis is the Bcl-2 family, which includes both anti-apoptotic and pro-apoptotic members. The relative balance, rather than absolute levels, of these proteins determines a cell's fate.
Cancer cells often overexpress anti-apoptotic proteins (e.g., Bcl-2 in prostate cancer, Bcl-xL in drug-resistant tumors) or downregulate pro-apoptotic ones (e.g., Bax mutations in colorectal cancer), leading to reduced apoptosis and therapy resistance.[6]
5.1.2 p53
p53, known as "the guardian of the genome," is a tumor suppressor protein that regulates the cell cycle and apoptosis in response to DNA damage. The TP53 gene is mutated in over 50% of cancers, impairing apoptosis and enabling tumor progression.
5.1.3 Inhibitor of Apoptosis Proteins (IAPs)
IAPs are a family of proteins that block apoptosis by inhibiting caspases directly or promoting their degradation. They contain BIR domains that bind caspases and prevent cell death.
5.2 Reduced Caspase Activity
Caspases are critical enzymes involved in apoptosis and inflammation. In cancer, reduced expression or loss of caspase activity impairs apoptosis, allowing abnormal cells to survive, proliferate, and resist treatment—contributing to carcinogenesis.
Types of Caspases:
Clinical Evidence of Caspase Downregulation in Cancer:
5.3 Impaired Death Receptor Signaling
The extrinsic pathway of apoptosis is initiated by death receptors located on the cell surface, which respond to external death signals (ligands) like FasL, TNF, and TRAIL. These receptors include TNFR1, Fas (CD95), DR3, DR4, DR5, and others, each containing a death domain that activates downstream caspases to induce apoptosis.
Mechanism of Normal Death Receptor Function:
Mechanisms of Impairment in Cancer:
6.0 Targeting the Bcl-2 Family in Cancer Therapy
1. Antisense Inhibitors
2. Small Molecule Inhibitors
3. BH3 Mimetics
4. Gene Silencing
Impact: Increases cancer cell susceptibility to apoptosis and reduces therapy resistance.
6.1Targeting Apoptosis in Cancer Therapy
Apoptotic pathways, when disrupted, not only contribute to cancer development but also offer promising targets for treatment. Defects or abnormalities within these pathways can be exploited therapeutically—by designing drugs or interventions that restore normal apoptotic signaling, it becomes possible to selectively eliminate cancer cells that rely on such defects for survival. Recent advances in research have led to the discovery of novel anticancer agents that specifically target these apoptotic dysfunctions[14]. This section focuses on emerging treatment strategies aimed at correcting the apoptotic defects discussed earlier in Section 3. For a detailed overview of these drugs and approaches, please refer to Table 1.
|
Targeting the Bcl-2 family of proteins |
Remark |
|
Agents that target the Bcl-2 family proteins |
Oblimersen sodium |
|
Reported to show chemosensitising effects in combined treatment with conventional anticancer drugs in chronic myeloid leukaemia patients and an improvement in survival in these patients |
|
|
Small molecule inhibitors of the Bcl-2 family of proteins |
|
|
Molecules reported to affect gene or protein expression include sodium butyrate, depsipetide, fenretinide and flavipirodo. Molecules reported to act on the proteins themselves include gossypol, ABT-737, ABT-263, GX15-070 and HA14-1 |
|
|
BH3 mimetics |
|
|
ABT-737 reported to inhibit anti-apoptotic proteins such as Bcl-2, Bcl-xL, and Bcl-W and to exhibit cytotoxicity in lymphoma, small cell lung carcinoma cell line and primary patient-derived cells |
|
|
ATF4, ATF3 and NOXA reported to bind to and inhibit Mcl-1 |
|
|
Silencing the Bcl family anti-apoptotic proteins/genes |
Bcl-2 specific siRNA reported to specifically inhibit the expression of target gene in vitro and in vivo with anti-proliferative and pro-apoptotic effects observed in pancreatic carcinoma cells |
|
Silencing Bmi-1 in MCF breast cancer cells reported to downregulate the expression of pAkt and Bcl-2 and to increase sensitivity of these cells to doxorubicin with an increase in apoptotic cells in vitro and in vivo |
|
|
Targeting p53 |
|
|
p53-based gene therapy |
First report on the use of a wild-type p53 gene containing retroviral vector injected into tumour cells of non-small cell lung carcinoma derived from patients. The use of p53-based gene therapy was reported to be feasible. |
|
Introduction of wild type p53 gene reported to sensitise tumour cells of head and neck, colorectal and prostate cancers and glioma to ionising radiation |
|
|
Genetically engineered oncolytic adenovirus, ONYX-015 reported to selectively replicate in and lyse tumour cells deficient in p53 |
|
|
p53-based drug therapy |
Small molecules |
|
Phikan083 reported to bind to and restore mutant p53 |
|
|
CP-31398 reported to intercalate with DNA and alter and destabilise the DNA-p53 core domain complex, resulting in the restoration of unstable p53 mutants |
|
|
Other agents |
|
|
Nutlins reported to inhibit the MSM2-p53 interaction, stabilise p53 and selectively induce senescence in cancer cells |
|
|
MI-219 reported to disrupt the MDM2-p53 interaction, resulting in inhibition of cell proliferation, selective apoptosis in tumour cells and complete tumour growth inhibition |
|
|
Tenovins reported to decrease tumour growth in vivo |
|
|
p53-based immunotherapy |
Patients with advanced stage cancer given vaccine containing a recombinant replication-defective adenoviral vector with human wild-type p53 reported to have stable disease |
|
Clinical and p53-specific T cell responses observed in patients given p53 peptide pulsed dendritic cells in a phase I clinical trial |
|
|
Targeting IAPS |
|
|
Targeting XIAP |
Antisense approach |
|
Reported to result in an improved in vivo tumour control by radiotherapy |
|
|
Concurrent use of antisense oligonucleotides and chemotherapy reported to exhibit enhanced chemotherapeutic activity in lung cancer cells in vitro and in vivo |
|
|
siRNA approach |
|
|
siRNA targeting of XIAP reported to increase radiation sensitivity of human cancer cells independent of TP53 status |
|
|
Targeting XIAP or Survivin by siRNAs sensitised hepatoma cells to death receptor- and chemotherapeutic agent-induced cell death |
|
|
Targeting Survivin |
Antisense approach |
|
Transfection of anti-sense Survivin into YUSAC-2 and LOX malignant melanoma cells reported to result in spontaneous apoptosis |
|
|
Reported to induce apoptosis and sensitise head and neck squamous cell carcinoma cells to chemotherapy |
|
|
Reported to inhibit growth and proliferation of medullary thyroid carcinoma cells |
|
|
siRNA approach |
|
|
Reported o downregulate Survivin and diminish radioresistance in pancreatic cancer cells |
|
|
Reported to inhibit proliferation and induce apoptosis in SPCA1 and SH77 human lung adenocarcinoma cells |
|
|
Reported to suppress Survivin expression, inhibit cell proliferation and enhance apoptosis in SKOV3/DDP ovarian cancer cells |
|
|
Reported to enhance the radiosensitivity of human non-small cell lung cancer cells |
|
|
Other IAP antagonists |
Small molecules antagonists |
|
Cyclin-dependent kinase inhibitors and Hsp90 inhibitors and gene therapy attempted in targeting Survivin in cancer therapy |
|
|
Cyclopeptidic Smac mimetics 2 and 3 report to bind to XIAP and cIAP-1/2 and restore the activities of caspases- 9 and 3/-7 inhibited by XIAP |
|
|
SM-164 reported to enhance TRAIL activity by concurrently targeting XIAP and cIAP1 |
|
|
Targeting caspases |
|
|
Caspase-based drug therapy |
Apoptin reported to selectively induce apoptosis in malignant but not normal cells |
|
Small molecules caspase activators reported to lower the activation threshold of caspase or activate caspase, contributing to an increased drug sensitivity of cancer cells |
|
|
Caspase-based gene therapy |
Human caspase-3 gene therapy used in addition to etoposide treatment in an AH130 liver tumour model reported to induce extensive apoptosis and reduce tumour volume |
|
Gene transfer of constitutively active caspse-3 into HuH7 human hepatoma cells reported to selectively induce apoptosis |
|
|
A recombinant adenovirus carrying immunocaspase 3 reported to exert anticancer effect in hepatocellular carcinoma in vitro and in vivo |
6.2 Targeting p53
6.2.1 p53-Based Gene Therapy
6.2.2 p53-Based Drug Therapy
6.2.3 p53-Based Immunotherapy
6.3 Introduction to IAPs
Inhibitor of Apoptosis Proteins (IAPs) are a family of proteins that prevent cell death by blocking caspases, which are essential for the execution of apoptosis. Their overexpression in cancer cells contributes to therapy resistance and tumor survival, making them a key target in cancer therapy.
6.3.1 XIAP Targeting Approaches
XIAP (X-linked inhibitor of apoptosis protein) is the most potent caspase inhibitor in the IAP family, directly blocking caspase-9, -3, and -7. Therapeutic strategies such as antisense oligonucleotides and siRNAs have been developed to silence XIAP expression. These approaches enhance the effectiveness of radiation and chemotherapy in various cancers, including liver and p53-deficient tumors, by sensitizing the cells to undergo apoptosis.
6.3.2 Targeting Survivin
Survivin is another major IAP family member, widely overexpressed in many types of cancer. It supports tumor cell survival and therapy resistance. Antisense oligonucleotides targeting Survivin induce apoptosis in cancers like melanoma, head and neck, and thyroid. Similarly, siRNA-mediated inhibition of Survivin sensitizes pancreatic, lung, ovarian, and other cancers to radiotherapy and chemotherapy. Small-molecule inhibitors such as CDK inhibitors and Hsp90 inhibitors also suppress Survivin function, promoting cancer cell death.[10]
6.3.4 Use of Smac/DIABLO Mimetics
Smac/DIABLO mimetics are designed to imitate natural IAP inhibitors and restore caspase activity. These compounds target XIAP and other IAPs like cIAP1 to promote apoptosis. Examples include Smac mimetics “2” and “3,” and a non-peptidic Smac mimetic called SM-164. SM-164 inhibits both XIAP and cIAP1, enhancing TRAIL-induced apoptosis in cancer cells. These agents hold great promise in restoring the apoptotic capacity of cancer cells and improving treatment outcomes.
6.4 Targeting Caspases
Caspases, essential enzymes that execute apoptosis, are being actively explored as therapeutic targets in cancer treatment. Two main strategies—drug therapy and gene therapy—aim to restore caspase activity and promote cancer cell death. In caspase-based drug therapy, agents like Apoptin have shown the ability to induce apoptosis specifically in cancer cells without harming normal ones. Additionally, small molecules containing the RGD motif can directly activate procaspase-3, enhancing the apoptotic response and improving the efficacy of chemotherapy. Gene therapy approaches involve delivering caspase genes directly into tumors. For instance, introducing the caspase-3 gene in combination with chemotherapy in liver tumor models led to greater tumor reduction than chemotherapy alone. Similarly, using constitutively active caspase-3 in liver cancer cells or employing recombinant adenoviruses carrying immunocaspase-3 has proven effective in inducing targeted apoptosis in cancer cells, both in vitro and in vivo.
6.5 Molecules Targeting Apoptosis in Clinical Trials
Numerous apoptosis-modulating compounds have entered clinical trials, as documented in global registries like ClinicalTrials.gov. These investigational agents primarily target crucial apoptotic regulators, including members of the Bcl-2 family and IAPs. Their ongoing clinical evaluation reflects growing interest in leveraging apoptosis restoration as a viable cancer treatment strategy[11].
7.0 CONCLUSION
Research has shown that abnormalities in apoptotic pathways are crucial contributors to cancer development. Promisingly, new therapies that aim to restore or manipulate apoptosis are emerging, offering hope for more effective cancer treatments. Some of these therapies are in early experimental stages, while others have progressed to clinical trials, often being tested alongside traditional cancer drugs to enhance their effectiveness. Despite these advances, several concerns remain unresolved. There is uncertainty about whether tumors might develop resistance to apoptosis-targeting treatments, and whether these therapies might harm healthy cells. These concerns are significant given the known side effects and resistance issues associated with conventional chemotherapy, which tends to affect both cancerous and normal cells indiscriminately. Although the ideal treatment would selectively target a single apoptotic protein or pathway to reduce damage to healthy tissue, most current drugs—such as Bcl-2 inhibitors and IAP antagonists—affect multiple targets. This complexity underscores the need for long-term follow-up studies to assess the true outcomes for patients. Future research should focus on developing treatments that specifically induce apoptosis in cancer cells while sparing normal cells, thereby increasing the effectiveness of therapy and reducing side effects.
REFERENCES


Ajeet Kumar*, Sana Nusrat Praween, A Review on ‘’Apoptosis in Cancer: From Pathogenesis to Cancer’’, Int. J. of Pharm. Sci., 2025, Vol 3, Issue 9, 1953-1965 https://doi.org/10.5281/zenodo.17149896
 10.5281/zenodo.17149896
10.5281/zenodo.17149896
