
We use cookies to ensure our website works properly and to personalise your experience. Cookies policy
Department of Pharmaceutics, Kamalakshi Pandurangan College of Pharmacy, Ayyampalayam, Tiruvannamalai - 606 603.
Developing a new drug requires a great amount of research work in chemistry, manufacturing, controls, preclinical science, and clinical trials. Drug reviewers in regulatory agencies around the world bear the responsibility of evaluating whether the research data support the safety, effectiveness, and quality control of a new drug product to serve the public health. Every country has its own regulatory authority, which is responsible for enforcing the rules and regulations and issuing the guidelines to regulate the marketing of the drugs. This article focuses on the drug approval process in the USFDA.
American consumers benefit from having access to the safest and most advanced pharmaceutical system in the world. The main consumer watchdog in this system is the FDA's Centre for Drug Evaluation and Research (CDER). The centre’s best-known job is to evaluate new drugs before they can be sold. CDER's evaluation not only prevents quackery but also provides doctors and patients with the information they need to use medicines wisely. The centre ensures that drugs, both brand-name and generic, work correctly and that their health benefits outweigh their known risks. Drug companies seeking to sell a drug in the United States must first test it. The company then sends CDER the evidence from these tests to prove the drug is safe and effective for its intended use. A team of CDER physicians, statisticians, chemists, pharmacologists, and other scientists reviews the company's data and proposed labelling. If this independent and unbiased review establishes that a drug's health benefits outweigh its known risks, the drug is approved for sale. The centre doesn't actually test drugs itself, although it does conduct limited research in the areas of drug quality, safety, and effectiveness standards. Before a drug can be tested in people, the drug company or sponsor performs laboratory and animal tests to discover how the drug works and whether it's likely to be safe and work well in humans. Next, a series of tests in people is begun to determine whether the drug is safe when used to treat a disease and whether it provides a real health benefit.[2]

Figure 01: The basic Regulation [3]
Investigational New Drug (IND) Application
Current Federal law requires that a drug be the subject of an approved marketing application before it is transported or distributed across state lines. Because a sponsor will probably want to ship the investigational drug to clinical investigators in many states, it must seek an exemption from that legal requirement. The IND is the means through which the sponsor technically obtains this exemption from the FDA. During a new drug's early preclinical development, the sponsor's primary goal is to determine if the product is reasonably safe for initial use in humans, and if the compound exhibits pharmacological activity that justifies commercial development. When a product is identified as a viable candidate for further development, the sponsor then focuses on collecting the data and information necessary to establish that the product will not expose humans to unreasonable risks when used in limited, early-stage clinical studies. FDA's role in the development of a new drug begins when the drug's sponsor (usually the manufacturer or potential marketer), having screened the new molecule for pharmacological activity and acute toxicity potential in animals, wants to test its diagnostic or therapeutic potential in humans. At that point, the molecule changes in legal status under the Federal Food, Drug, and Cosmetic Act and becomes a new drug subject to specific requirements of the drug regulatory system. There are three IND types:
There are two IND categories:
The IND application must contain information in three broad areas:
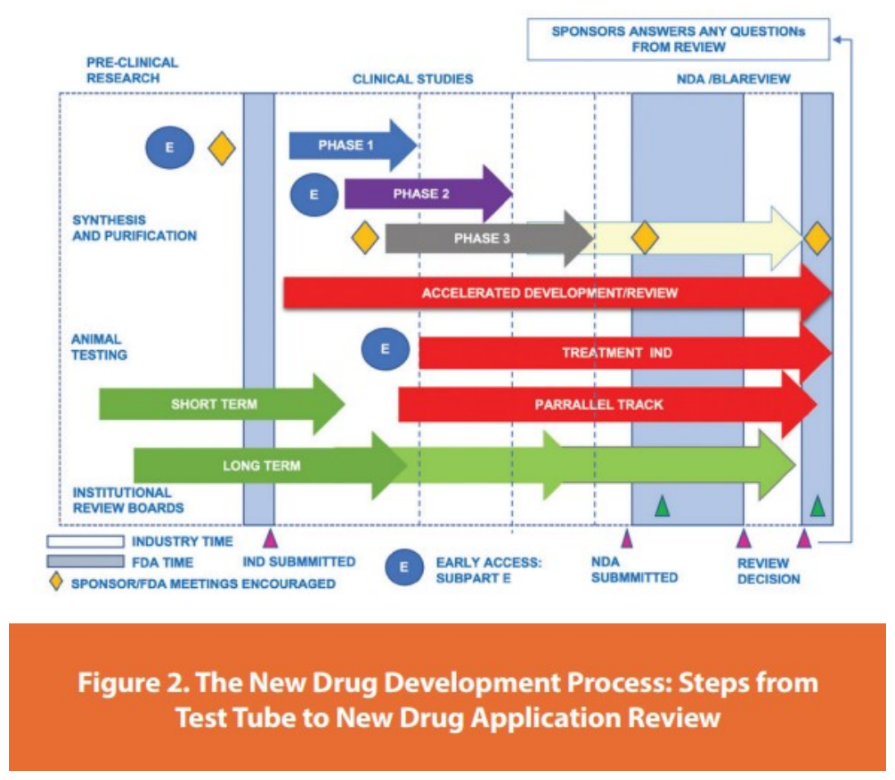
Figure No. 02: Drug approval process [5]
4. New Drug Application (NDA)
Submitting a New Drug Application (NDA) to the U.S. Food and Drug Administration (FDA) is a significant milestone in drug development, marking the final step before a drug can reach patients. It represents the culmination of years of research, preclinical studies, clinical trials, and regulatory planning, all designed to demonstrate a drug’s safety, efficacy, and manufacturing quality. A well-prepared NDA submission is essential for a smooth FDA review, helping to prevent delays and requests for additional data. Under the Food, Drug, and Cosmetic Act, all new drugs, biologics, and therapies must undergo rigorous evaluation before they can be marketed. The NDA compiles clinical trial data, pharmacology and toxicology summaries, proposed labeling, and manufacturing details to confirm that a drug’s benefits outweigh its risks. For pharmaceutical companies and scientists, understanding the NDA process, regulatory expectations, and FDA review steps is key to securing timely approval. The NDA follows the Electronic Common Technical Document (eCTD) format, which organizes key data into five structured modules for evaluation by the Center for Drug Evaluation and Research (CDER).
This guide provides a breakdown of the NDA process:
Why are NDAs Important?
NDAs play a crucial role in ensuring patient safety and drug effectiveness. We discussed before that a regulatory submission is like the identification of any medicinal product, telling its story from creation to side effects. From A to Z. NDA plays the same role for new medicines that are planned to be marketed for the very first time. By thoroughly reviewing the information in an NDA, regulatory authorities assess the potential benefits and risks of a new drug before it’s available for public use.[7]
Importance of NDA Submissions in Pharmaceutical Development
NDAs are the cornerstone of bringing safe and effective new drugs to market. In other words, if a new drug is being marketed for the very first time, an NDA submission is required. Depending on where the drug will be marketed, an NDA needs to be submitted to the respective health authority of that region. For example, the New Drug Application (NDA) has been the cornerstone of ensuring safe and effective medications reach American patients for over eight decades. Since 1938, every new drug has undergone a rigorous NDA review process before hitting pharmacy shelves in the US. This in-depth evaluation by the FDA plays a vital role in protecting public health in the US. [8]
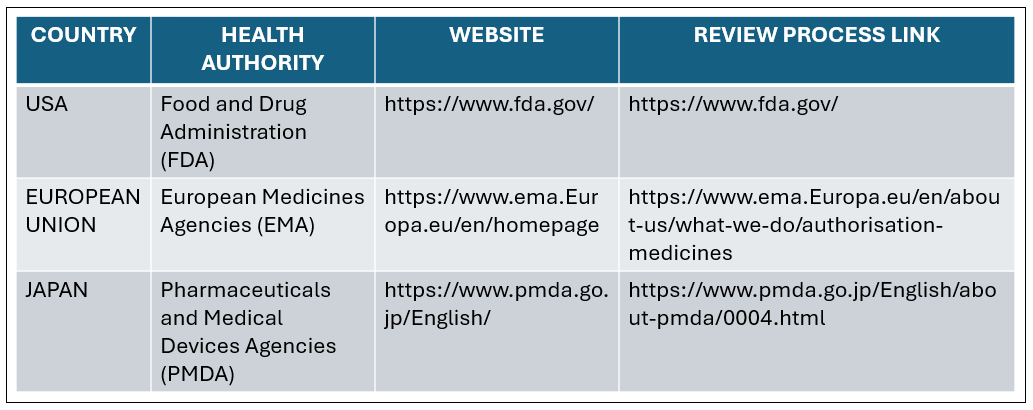
Regulatory bodies that are involved in various countries is shown in table no.01
Table 01: Regulatory Bodies involved in NDA Approvals [9]
NDA Forms and Electronic Submissions

Figure 04: U.S.FDA Drug approval process [11]
Drug Development Process
Step 01: Discovery And Development
Typically, researchers discover new drugs through:
At this stage in the process, thousands of compounds may be potential candidates for development as a medical treatment. After early testing, however, only a small number of compounds look promising and call for further study.
Development
Once researchers identify a promising compound for development, they conduct experiments to gather information on:
Step 02: Preclinical Research
Phases of preclinical research
Preclinical research is generally divided into four phases: basic research, drug discovery and candidate nomination, lead optimization and investigational new drug (IND)-enabling studies.
Phase 1
Basic research
Basic research comprises any studies conducted by academic institutions, pharmaceutical companies and biotechnology firms to understand the underlying biology of a disease and how that disease might be treated. This is often where scientists discover drug targets – the biological processes or pathways that play a role in a particular condition, which they will then attempt to modify with drugs to treat the disease. Once a target is identified, the next step is target validation, in which researchers gather evidence to confirm the therapeutic effects of target modulation. This can involve a range of techniques, such as genetic studies, biochemical assays and animal models.
Phase 2
Drug discovery and candidate nomination
After basic research and target validation are complete, the focus shifts to finding or designing molecules that can interact with the target in a specific and effective manner. In the drug discovery phase, researchers begin testing potential therapeutic compounds, narrowing down the possibilities from a nearly infinite number to one drug candidate for clinical testing. These experiments are often conducted in cellular models of a disease. The compounds that show promise in these tests are known as hits. In the drug candidate nomination process, various factors are considered when selecting the most promising of these for further development, including potency, selectivity, pharmacokinetics, safety profile and potential for formulation.

Phase 3
Lead optimization
Compounds that appear promising in the in vivo tests are called leads. As researchers learn more about which leads appear to work well, and which ones appear ineffective and/or harmful, they may chemically modify the compound in question to try to improve its performance. Through this stage of study, scientists will also gather information on which doses are the safest and most effective and build a dosing strategy. This third stage of preclinical research is known as lead optimization, referring to the process of arriving at the best possible drug candidate.
Once a lead drug candidate is identified, a typical preclinical development program consists of six major segments:

Development of the dosage formulation
Proper dosing determines medication effectiveness. After researchers identify a promising compound for development, they conduct experiments to gather information on:

Figure 05: The six steps of new drug development towards FDA approval [14]
Step 03: Clinical Research
Phase O trials
First-in-human clinical trial (optional) with 10 to 15 healthy volunteers where small amounts of the investigational drug are given to check if the drug behaves as expected in humans. If the medication acts differently than expected, additional preclinical research will most likely be completed before deciding whether to continue.
Phase I trials
Researchers test a drug or treatment in 20 to 80 people (many times a first-in-human study) that are often healthy volunteers to see if the drug or treatment is safe, identify side effects, and determine safe dosage ranges. Recruiting patients for this phase often involves finding healthy volunteers willing to go through rigorous testing while staying at the research facility.
Phase II trials
The investigational drug is given to a larger group (hundreds) of people who have the disease of interest. This is to determine its effectiveness and to further study safety, short-term side effects, and dosages. Sometimes phase 2 studies are divided into phases 2a and 2b:
Participants for phase II research need to have the target disease but not a lot of comorbidities. Inclusion and exclusion criteria are more restrictive, making recruitment a challenge. A targeted recruitment plan is needed.
Phase III trials
The investigational drug or treatment is given to large groups of people (thousands) to confirm its effectiveness, monitor side effects, compare it with standard or similar treatments, and collect information that will allow the new drug or treatment to be used safely. Sometimes phase 3 studies are done as 3a and 3b:
Phase III study Inclusion and exclusion criteria are less restrictive, but a large number of patients are needed, often from several countries. A strong global recruitment plan is needed.[15]
Phase 4 trials
After these phases, a medicine reaches Phase 4, which occurs after it receives regulatory approval. Phase IV studies (also known as post-marketing surveillance trials) play a key role in the clinical research continuum.
The purpose of Phase 4 clinical trials
Hundreds of thousands of registered clinical studies are conducted worldwide every year. After the first three phases of a drug trial, a Phase IV study is conducted to collect even more information about how well a new treatment works, its safety among a larger number of patients, and its outcomes over a longer period. Sometimes, this phase also looks at how the drug works in patients with certain characteristics or to compare/combine the treatment with other drugs. The goal of these trials is to conduct ongoing safety surveillance, identify rare adverse reactions or harmful effects, assess efficacy, and optimise the drug's use. Phase 4 clinical trials also play a big role in responding to regulatory requirements (if necessary) and supporting label expansions or new indications that might come to light.
Conducting a Phase 4 clinical trial
Phase 4 trials are often conducted as observational studies, so researchers don’t actively control the treatment participants receive. Instead, they observe how patients take the medication according to the current medical practice. The idea is for researchers to exert as little influence as possible and to study the medication under real-life conditions. Other types of Phase 4 clinical trials include randomised controlled trials. In this type of trial, participants are randomly assigned to an experiment group (which receives the medication being tested) and a control group. There are also registries, which are observational and record patient data in real-world settings. When designing and executing a Phase 4 clinical trial, it’s crucial that the trial results in valuable and actionable efficacy data.
To achieve this, considerations might include:
Key components of Phase 4 trials
Phase 4 clinical trials have several key components, including pharmacovigilance. Also called PV, this refers to a drug safety reporting system that monitors drug effects, keeping a particularly close eye on suspected adverse drug reactions or safety “signals,” which can include rare side effects. Other aspects of Phase 4 clinical trials include real-world evidence and a broad patient population. By observing patients like these under normal clinical use conditions, clinical trials gather evidence essential for understanding a drug's long-term efficacy and implications. As with all trials, Phase 4 trials have eligibility and exclusion criteria that determine who can participate in the study.
Challenges and considerations
Executing successful, safe, and accurate Phase 4 clinical trials can present unique challenges. For example, recruiting a large and diverse patient population can be difficult. Likewise, maintaining patient compliance (particularly over long periods of time) can be a hurdle. Managing and analysing the often-massive datasets generated by real-world studies can also be challenging. This requires sophisticated techniques and systems to ensure data is stored, assessed, and acted on appropriately.
Regulatory oversight and ethical considerations
Phase 4 clinical trials have many regulatory requirements and ethical considerations specific to post-marketing research. Firstly, regulatory authorities play a critical role in overseeing these trials. They ensure these trials adhere to strict ethical and scientific guidelines, protecting participants from risk. Like earlier trials, informed consent is a cornerstone of ethical research in Phase 4 clinical trials. Patients must be fully informed about the study’s objectives, potential risks and benefits, and their right to withdraw at any time. Other ethical considerations include patient welfare, respect, participant privacy, and data confidentiality (which must be meticulously protected throughout the study).
Examples of Phase 4 clinical trials
Phase 4 clinical trials can significantly impact medical practice and patient care. For a recent success story, we can look at the results from a Phase 4 trial with CHIMES, announced by Genentech (a member of the Roche Group). This trial was the first-ever clinical trial conducted exclusively in Black and Hispanic/Latinx people with relapsing multiple sclerosis. Importantly, it set a new standard for inclusive research in MS. [16]
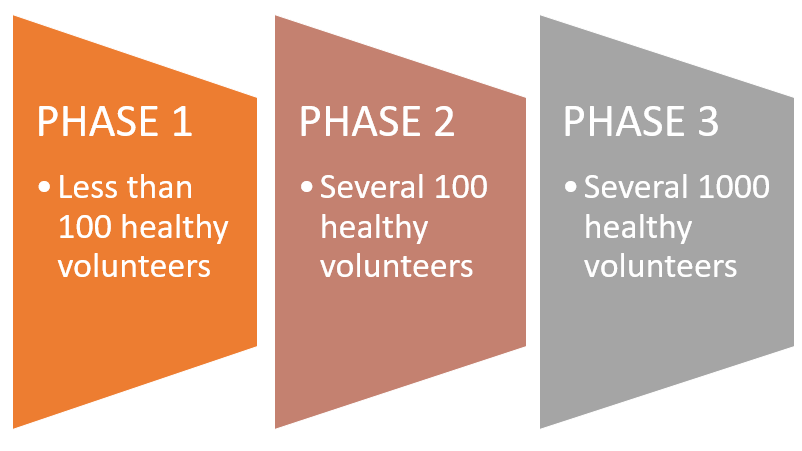
Figure 06: Three phases of clinical trials [17]
Step 04: FDA Drug Review
FDA Review and Approval
After completion of the first 3 phases of clinical trials, the Sponsor analyzes the data. If the data shows that the therapy is effective and safe, the company submits a New Drug Application (NDA) to the FDA. Your group may wish to become a member of the FDA Advisory Committee if one is needed during the review process. An NDA will contain:
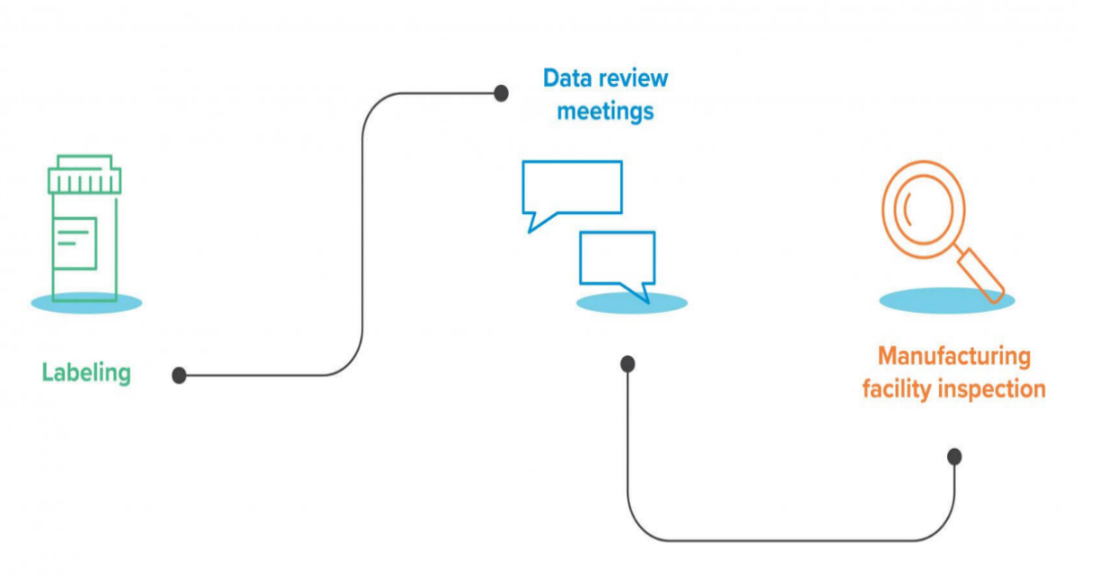
Figure 07: NDA Review process [19]
Drug labelling:
The agency reviews the labelling to ensure that it adequately and accurately communicates necessary information about the drug to health care professionals and consumers. This review process also verifies that false claims are not being made in the labelling of all end products.
NDA review/application review and sponsor meetings:
This includes a multi-step process that incorporates detailed reviews of the sponsor’s research, clinical safety data, animal and human analysis data, as well as meetings with the sponsor.
Manufacturing facility inspection:
Prior to the final approval, the FDA will inspect the manufacturing facility to ensure it’s compliant with current Good Manufacturing Practices (cGMP) and it’s safe. After a drug successfully completes all these steps, the FDA grants drug approval and the drug product can be supplied to the market. Additionally, there is a formal fourth phase: a Post Marketing and Post Approval Risk Assessment. This monitors the drug’s safety after it is on the market and detects any serious, unexpected adverse events that were not possible to predict during the clinical trials. The sponsor submits periodic updates to the FDA. [20]
Step 05: FDA Post-Market Drug Safety Monitoring
Even though clinical trials provide important information on a drug’s efficacy and safety, it is impossible to have complete information about the safety of a drug at the time of approval. Despite the rigorous steps in the process of drug development, limitations exist. Therefore, the true picture of a product’s safety actually evolves over the months and even years that make up a product’s lifetime in the marketplace. FDA reviews reports of problems with prescription and over-the-counter drugs, and can decide to add cautions to the dosage or usage information, as well as other measures for more serious issues.
On this page you will find information on:
Supplemental Applications
Developers must file a supplemental application if they wish to make any significant changes from the original NDA. Generally, any changes in formulation, labeling, or dosage strength must be approved by FDA before they can be made.
INDs for Marketed Drugs
If sponsors want to further develop an approved drug for a new use, dosage strength, new form, or different form (such as an injectable or oral liquid, as opposed to tablet form), or if they want to conduct other clinical research or a post-market safety study, they would do so under an IND.
Manufacturer Inspections
FDA officials conduct routine inspections of drug manufacturing facilities across the United States, and abroad if approved products are manufactured overseas. Manufacturers may be informed of inspections in advance, or the inspections may be unannounced. Inspections may be routine or caused by a particular problem or concern. The purpose of these inspections is to make sure that developers are following good manufacturer practice. FDA can shut down a facility if minimum standards are not met.
Drug Advertising
FDA regulates prescription drug advertisements and promotional labelling. By law, a developer is prohibited from advertising unapproved uses of their product. All advertisements, such as product claims or reminder ads, cannot be false or misleading. They must contain truthful information about a drug’s effectiveness, side effects, and prescribing information. These advertisements can be found in medical journals, newspapers, and magazines, and on the Internet, television, or radio. Promotional labelling differs from drug advertisements in the way it is distributed. Pharmaceutical companies give out brochures or other promotional materials to physicians or consumers. The drug’s prescribing information must accompany promotional labelling. Learn more at Prescription Drug Advertising.
Generic Drugs
New drugs are patent protected when they are approved for marketing. This means that only the sponsor has the right to market the drug exclusively. Once the patent expires, other drug manufacturers can develop the drug, which will be known as a generic version of the drug. Generic drugs are comparable to brand name drugs and must have the same:
Because generic drugs are comparable to drugs already on the market, generic drug manufacturers do not have to conduct clinical trials to demonstrate that their product is safe and effective. Instead, they conduct bio-equivalence studies and file an Abbreviated New Drug Application. Learn more at Generic Drugs: Questions and Answers.
Reporting Problems
The FDA has several programs that allow manufacturers, health professionals, and consumers to report problems associated with approved drugs.
Active Surveillance
Under the Sentinel Initiative, the FDA is developing a new national system to more quickly spot possible safety issues. The system will use very large existing electronic health databases—like electronic health records systems, administrative and insurance claims databases, and registries—to keep an eye on the safety of approved medical products in real time. This tool will add to, but not replace, the FDA's existing post-market safety assessment tools. Learn more about the Sentinel Initiative and its major activities.[21]
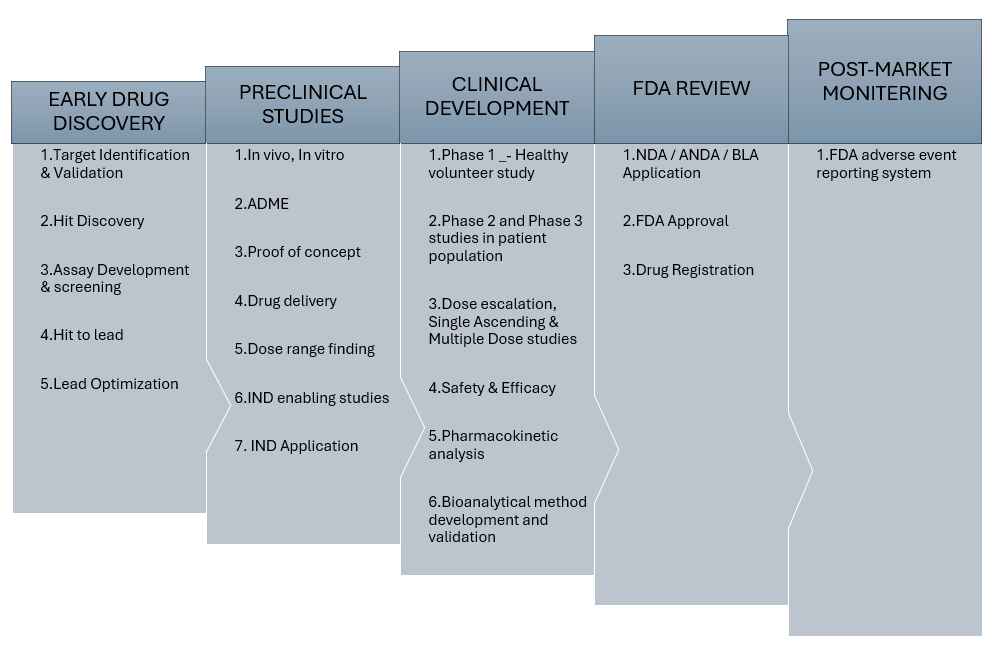
Figure 08: Flow chart of drug development process [22]
5. CONCLUSION
In conclusion, understanding the FDA approval process is essential for anyone involved in the pharmaceutical industry. From preclinical testing to clinical trials and the NDA review, each step plays a vital role in ensuring the safety and efficacy of drugs and medical devices. By partnering with experienced CROs like Lindus Health, pharmaceutical companies can navigate this complex process with confidence, knowing that they have the support and expertise necessary for success.
ACKNOWLEDGEMENT:
We express our sincere gratitude to the management of Kamalakshi Pandurangan college of Pharmacy, particularly Dr. D. Rajalingam, Principal, Dr. N. Gnanasekar, Vice Principal, Dr. V. Kannabirran Professor cum Head, Mr. K. Senthil Kumar, Associate Professor, Mrs. M. Bharathi, Associate Professor, Department of Pharmaceutics, faculty, and non-teaching staff for their guidance invaluable support and assistance in the successful completion of our project.
REFERENCES



Gilbert Tony K., Pavithra R., Arasu T., Mahendiran K., Bharathi M.*, Kannabirran V., Rajalingam D., Gnanasekar N., A Review on Drug Approval Process in USFDA, Int. J. of Pharm. Sci., 2025, Vol 3, Issue 8, 2275-2291 https://doi.org/10.5281/zenodo.16924714
 10.5281/zenodo.16924714
10.5281/zenodo.16924714