Delonix Society's Baramati College of pharmacy, Barhanpur.
The conventional drug development process has a high attrition rate, is expensive, and takes a long time. Machine learning (ML) and artificial intelligence (AI) have become revolutionary technologies that can find new therapeutic candidates, increase efficiency, and lower prices. Supervised and unsupervised learning, deep learning (CNNs, RNNs), graph neural networks (GNNs), natural language processing (NLP), generative/reinforcement learning models, and their applications throughout the drug discovery process are all covered in detail in this review. We look at these techniques in the areas of drug repurposing, ADMET prediction, virtual screening and lead optimization, target discovery, and clinical trial design. Important topics are covered, including the availability and quality of data, the interpretability of models, ethical and legal concerns, and the requirement for experimental integration. We focus on upcoming topics like explainable AI, hybrid computational-experimental techniques, and frontier technologies like quantum computing. Finally, recent commercial examples demonstrate real-world applications of AI in drug discovery. Given the rapid evolution of AI/ML, ongoing innovation and collaboration will be required to fully fulfill their promise to alter pharmaceutical research and development.
The process of medication discovery and development has typically been long, difficult, and costly, frequently requiring over a decade of research and billions of dollars before a novel therapy reaches the market. The intrinsic complexity of biological systems, the size of chemical space, and the high attrition rates at different phases of preclinical and clinical research are the causes of these difficulties. However, artificial intelligence (AI) and its subset machine learning (ML) have emerged in recent years as revolutionary technologies that have the potential to overcome many of these constraints by facilitating quicker, more intelligent, and more effective decision-making throughout the drug discovery process. AI/ML systems can learn patterns from large, multidimensional datasets, such as chemical structures, biological assays, genomics, and clinical data, and then use these patterns to generate predictive models that can speed up key stages like target identification, virtual screening, lead optimization, and toxicity prediction.[1]
The use of AI/ML in pharmaceutical research is indicative of a larger trend toward data-driven innovation, in which computational techniques supplement traditional experimental techniques. Deep learning, graph neural networks, natural language processing, and generative models have all been employed to solve important difficulties in drug design, enhancing hit rates and reducing trial burden. Such techniques are not only boosting the efficiency of traditional workflows but also enabling whole new tactics — for example, de novo molecular creation and real-time integration of multi-omics data.[2]
Despite encouraging advances, successfully integrating AI/ML into drug research presents obstacles. Data quality and availability, model interpretability, ethical considerations, and regulatory compliance are all ongoing challenges that must be addressed before these technologies can reach their full potential. Furthermore, the seamless integration of AI predictions with experimental validation is still a topic of ongoing study and practical concern.[3]
In this comprehensive review, we synthesize the most recent advances in AI and ML methodologies used in drug discovery, examine their impact on various stages of the drug development process, and discuss the key challenges and future directions that will shape the next generation of AI-enabled pharmaceutical innovation.
BACKGROUND / HISTORICAL PERSPECTIVE:
The incorporation of computational tools into drug discovery began decades before the contemporary increase in artificial intelligence (AI) and machine learning (ML) technology. Early computational approaches centered on computer-aided drug design (CADD), which employed basic statistical and mathematical tools to link chemical structures to biological activity. Quantitative structure-activity relationship (QSAR) models, which were originally constructed with linear regression and simple descriptors, assisted scientists in predicting how changes in molecular structures will alter biological function, allowing for more reasonable compound selection for experimental testing. These methods were frequently used with molecular docking simulations to determine the fit between prospective drug compounds and target proteins, representing the initial steps toward in silico drug creation. Classical approaches like QSAR and docking laid the foundation for computational screening long before advanced AI systems were feasible.[4]
As processing power grew and massive chemical and biological information became accessible, the development toward AI/ML approaches quickened. In the late 1990s and early 2000s, traditional machine learning algorithms such as support vector machines (SVMs), random forests, and k-nearest neighbors began to outperform linear statistical models in QSAR and bioactivity prediction tasks by capturing complex, nonlinear relationships in high-dimensional data. These ML methods enabled more robust models for ligand-target interactions, toxicity prediction, and early-stage virtual screening, gradually replacing previous rule-based procedures with data-driven models.
The development of deep learning and sophisticated neural network topologies during the past ten years marked the real shift toward modern AI. Models can now learn molecular features directly from raw representations like SMILES strings and molecular graphs without the need for manual feature engineering thanks to deep learning techniques like convolutional neural networks (CNNs), recurrent neural networks (RNNs), and more recently graph neural networks (GNNs). These methods have greatly increased the predicted accuracy of models for molecular characteristics, bioactivity, and de novo drug discovery. By proposing completely new molecular structures with desired properties, generative models like variational autoencoders (VAEs) and generative adversarial networks (GANs) have significantly increased possibilities. The use of natural language processing (NLP) in mining biomedical literature and patents has also become critical for uncovering novel drug-target relationships and repurposing opportunities.
Over the past 20 years, research activity and publications in AI-driven drug development have increased dramatically, reflecting this shift. Studies involving ML and medicinal chemistry have grown dramatically since the 2000s, according to bibliometric assessments, with leading contributions from institutions all over the world in QSAR, virtual screening, and molecular design. Between 1990 and 2023, the number of articles published on AI applications in drug discovery increased significantly, demonstrating the field's overall maturity as well as the quick adoption of cutting-edge computational approaches.[5]
Collectively, this historical progression — from classical QSAR and docking to modern AI/ML frameworks — illustrates how drug discovery has shifted from rule-based, expert-driven processes to adaptive, data-driven systems capable of analyzing complex biological and chemical information at unprecedented scale. This evolution underpins the significant role that AI and ML now play in accelerating therapeutics development and shaping the future of pharmaceutical research.
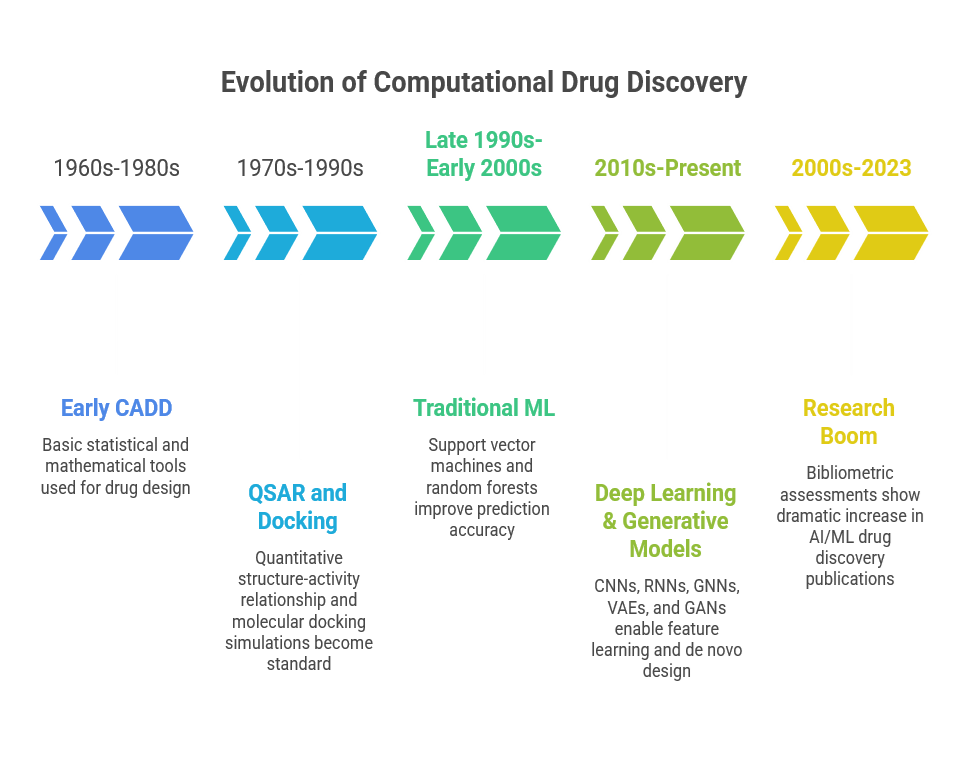
Figure 1: Evolution of Computational Drug Discovery[4,5]
CORE AI/ML METHODS USED IN DRUG DISCOVERY:
Complex biological, chemical, and clinical data can be analyzed using computational frameworks provided by artificial intelligence (AI) and machine learning (ML). By facilitating compound creation, prediction, and optimization as well as effective decision-making throughout the drug development process, these techniques have completely transformed drug discovery. The main AI/ML techniques frequently employed in pharmaceutical research are covered below.
1. Machine Learning (ML):[6]
Algorithms used in machine learning are able to recognize patterns in data and generate predictions without explicit programming. For the analysis of organized chemical and biological datasets, machine learning is especially helpful.
a) Supervised Learning:
Applications:
b) Unsupervised Learning:
Applications:
2. Deep Learning (DL):[7]
A branch of machine learning called "deep learning" makes use of multi-layered neural networks to extract hierarchical features from unprocessed data.
a) Convolutional Neural Networks (CNNs): Initially used for image processing, CNNs are now used for protein-ligand interactions, molecular graphs, and three-dimensional structures.
b) Recurrent Neural Networks (RNNs):
Applications:
3. Graph Neural Networks (GNNs):[8]
Atoms serve as nodes and bonds as edges in a natural graph representation of molecules. GNNs use this model to efficiently capture chemical and structural information.
4. Natural Language Processing (NLP):[9]
NLP has important uses in drug development and makes it possible for machines to comprehend and analyze human language.
5. Reinforcement Learning (RL) & Generative Models:[10]
Beyond predicting tasks, RL and generative AI are applied to molecular synthesis and optimization.
a) Generative Adversarial Networks (GANs): The discriminator assesses the chemical correctness of the new molecules produced by the generator. • Uses include scaffold optimization and de novo molecule synthesis.
b) Variational Autoencoders (VAEs): Acquire knowledge of a latent space for molecules and use sampling to create new compounds.
c) Reinforcement Learning (RL): Models are trained to produce compounds that are optimized for various goals (activity, toxicity, ADMET).
Summary:
Table 1: Summary of Core AI/ML Methods Used in Drug Discovery[6,7,8,9,10]
|
Method |
Key Feature |
Applications |
|
ML (RF, SVM) |
Structured prediction |
QSAR, virtual screening, ADMET |
|
DL (CNN, RNN) |
Automatic feature learning |
Property prediction, de novo design |
|
GNN |
Graph-based molecular modeling |
Structure-aware prediction, molecule generation |
|
NLP |
Text mining and knowledge extraction |
Literature/patent mining, drug repurposing |
|
GAN, VAE, RL |
Molecular generation & optimization |
Novel molecule design, multi-objective lead optimization |
APPLICATIONS ACROSS DRUG DISCOVERY PROCESS:
By facilitating improved data interpretation, quicker predictions, and more economical decision-making, artificial intelligence (AI) and machine learning (ML) are revolutionizing every phase of the drug discovery process. Below, we discuss key applications in major phases of drug discovery
1. Target Identification:[11]
The first important step in the drug discovery process is target identification. It entails figuring out which biological molecules typically proteins, genes, or pathways are connected to disease mechanisms and can be altered by possible treatments.
By combining and evaluating massive, intricate datasets from transcriptomics, proteomics, genomics, and other multi-omics sources that would be difficult to manually analyze, AI speeds up this process. Based on anticipated druggability and therapeutic relevance, machine learning models can identify hidden patterns and correlations between diseases and molecular targets. To increase target selection confidence, contemporary AI frameworks can also incorporate data from biomedical literature and public sources. By concentrating efforts on the most promising biological targets, these skills aid in cutting down on the time and expense of experimental validation.
Key benefits:
2. Virtual Screening & Lead Optimization:[11]
Finding small compounds or biologics that efficiently interact with a target is the next stage once it has been discovered. Computational models are used in virtual screening to quickly assess thousands to billions of compounds:
Deep learning and sophisticated scoring functions that exceed old techniques have now improved the early use of traditional docking and QSAR models.
Key benefits:
3. ADMET Prediction[12]
To determine whether a chemical has appropriate drug-like qualities, ADMET (absorption, distribution, metabolism, excretion, and toxicity) is essential. Costly late-stage attrition is frequently the result of failures at this point.
By understanding intricate connections between molecular characteristics and biological behavior from sizable datasets that contain pharmacokinetic and toxicity annotations, AI enhances ADMET prediction. Compared to traditional statistical models, machine learning and deep learning techniques have demonstrated more accuracy in forecasting certain characteristics. Moreover, graph neural networks (GNNs) can effectively capture structural information to improve prediction quality for ADME and toxicity endpoints.
This lessens the need for experimental testing and focuses resources early in the process on candidates with superior safety and efficacy profiles.
Key benefits:
4. Drug Repurposing:[13]
Finding new therapeutic applications for already-approved medications is known as "drug repurposing," or "drug repositioning." Repurposing can significantly cut development time and expense compared to innovative medication creation because safety data for authorized pharmaceuticals are already established. By analyzing massive datasets of molecular activity, illness markers, and biological literature, AI is excellent at repurposing. AI can recommend current medications that might work well against novel targets or illnesses thanks to pattern recognition and predictive modeling. For instance, by connecting illness characteristics with medication activity profiles, AI-based research assisted in identifying Baricitinib, which was first used to treat rheumatoid arthritis, as a possible COVID-19 treatment. This method increases therapeutic choices for unmet medical needs while also speeding up the development schedule.
Key benefits:
5. Clinical Trial Design & Patient Stratification:[14]
Beyond discovery, AI continues to influence drug development by enhancing the planning and conduct of clinical trials. Clinical trials are frequently expensive, time-consuming, and prone to failure if trial criteria, endpoint definitions, or patient selection are not ideal.
By examining patient genomic profiles, electronic medical records, and trial data from the past, AI aids in trial design optimization by:
AI improves safety monitoring, reduces trial durations, and raises the chance of success by detecting biomarkers for toxicity or response and intelligently customizing clinical studies.
Key contributions:
In simple: From early biological understanding to the conduct of clinical trials, AI and ML are transforming contemporary drug research. AI improves the pace and likelihood of delivering novel treatments to patients by facilitating more precise forecasts and effective decision-making at several stages, including target selection, screening, ADMET profiling, repurposing, and trial design.
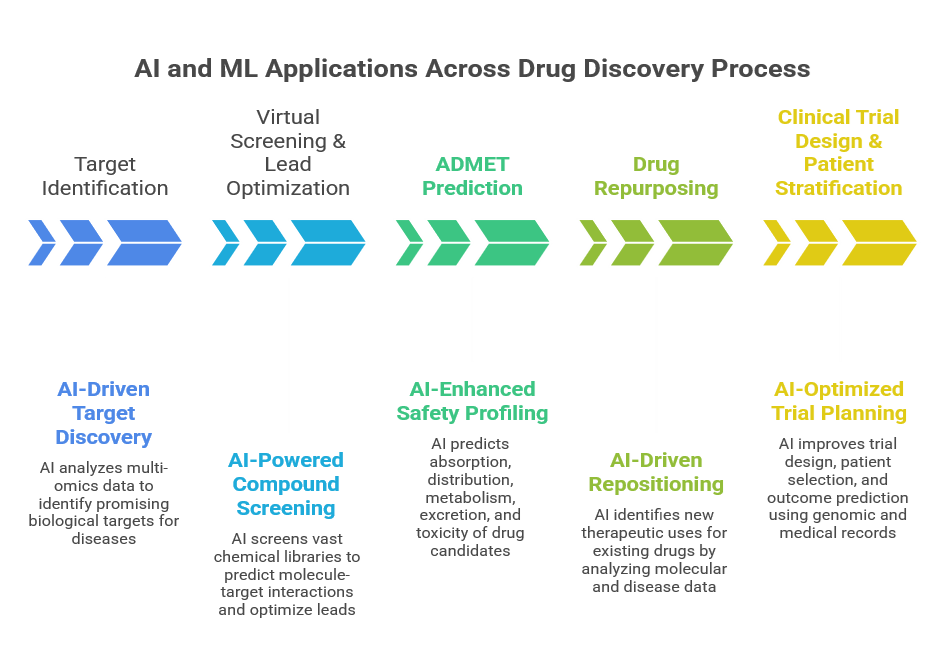
Figure 1: AI and ML Application Across Drug Discovery Process[11,12,13,14]
Table 2: Impact of AI on Efficiency and Cost Reduction[11,12,13,14]
|
Phase |
Traditional Approach |
AI-Driven Approach |
Key Advantage |
|
Target discovery |
Manual literature analysis |
Automated omics + NLP |
Faster target prioritization |
|
Screening |
Wet lab HTS |
In silico virtual screening |
Reduced cost & time |
|
Lead optimization |
Trial-and-error chemistry |
Predictive SAR & generation |
Higher hit quality |
|
ADMET |
Late experimental testing |
Early AI-based filtering |
Lower attrition |
|
Repurposing |
Serendipitous |
Data-driven predictions |
Faster approvals |
|
Clinical trials |
Broad patient groups |
Stratified patient cohorts |
Higher success rate |
CHALLENGES AND LIMITATIONS OF AI/ML IN DRUG DISCOVERY:
Drug development has been greatly accelerated by artificial intelligence (AI) and machine learning (ML), but putting these technologies into practice is not without its difficulties. For integration into pharmaceutical R&D to be successful, certain constraints must be addressed.
1. Availability and Quality of Data[15]
Large, high-quality datasets are crucial for the training and validation of AI and ML models. However, bias, incompleteness, and shortage are common problems with data in drug discovery:
2. Model Interpretability and the "Black Box" Problem[16]
Many artificial intelligence models, especially deep learning networks, operate as "black boxes," generating predictions that lack explicit justifications:
3. Regulatory and Ethical Concerns[17]
When AI is used in medication research, ethical and regulatory concerns are raised:
4. Integration with Experimental Validation[18]
An overview: AI and ML in drug development encounter significant obstacles pertaining to data quality, model openness, regulatory compliance, ethical issues, and practical experimental integration, despite their revolutionary promise. To fully achieve the promise of AI-driven drug development, these constraints must be addressed with improved datasets, explainable models, regulatory frameworks, and tight cooperation between computational and experimental teams.
EMERGING TRENDS & FUTURE DIRECTIONS IN AI/ML-DRIVEN DRUG DISCOVERY:[19]
The use of machine learning (ML) and artificial intelligence (AI) in drug discovery is developing quickly. Beyond existing uses, a number of new developments hold potential for further altering the pharmaceutical R&D environment.
1. Explainable AI (XAI) for Improved Trust and Interpretability
In AI-driven drug development, the "black box" nature of complex models such as deep learning networks is one of the main obstacles. The goal of explainable AI (XAI) techniques is to make model predictions understandable, actionable, and transparent:
Application: Interpreting QSAR and virtual screening predictions Creating compounds with the appropriate ADMET characteristics logically Drug target prioritization based on multi-omics data.
2. Hybrid Models Combining AI and Experimental Methods
Even if AI models can make accurate predictions, reliability still depends on experimental validation. Hybrid strategies that combine artificial intelligence with lab testing are becoming more popular:
Continuous learning procedures: AI forecasts compounds with promise → experimental tests confirm findings → fresh data feeds back into the model for improvement.
Multi-scale modeling: integrating high-throughput screening, biophysical simulations (such as quantum mechanics and molecular dynamics), and AI predictions.
Active learning frameworks: To enhance AI model performance, give priority to experiments that yield the most insightful data. Drug discovery and optimization campaigns are more likely to succeed when hybrid models bridge the gap between computational predictions and practical applicability.
3. Quantum Computing and Foundation Models
Emerging computational paradigms are expected to revolutionize AI-driven drug discovery:
a) Quantum Computing
b) Foundation Models[20]
Overview and Prospects for the Future:
Transparent, cooperative, and extremely predictive frameworks are the direction that AI in drug discovery is taking: Trust, interpretability, and regulatory compliance are guaranteed by explainable AI. Hybrid computational-experimental models lower attrition rates and improve forecast accuracy. In molecular simulations, prediction, and de novo drug creation, quantum computing and foundation models offer previously unheard-of speed, accuracy, and versatility. All of these developments point to AI actively changing the drug discovery process rather than just helping it, which will make it possible to produce next-generation treatments more quickly, safely, and affordably.
RECENT INDUSTRY & RESEARCH HIGHLIGHTS:
This section shows innovative real-world advancements that show how AI and ML technologies are increasingly boosting drugs discovery.
Large?Scale Biological Datasets and Consortium Initiatives:[21,22]
A significant industrial endeavor to produce the biggest single cell CRISPR perturbation dataset in the world, Illumina's Billion Cell Atlas is a ground-breaking tool for AI-driven drug development. Instead of depending only on literature or sparse experimental data, this dataset allows deep AI/ML models to understand actual biological reactions by capturing how 1 billion individual human cells react to genetic alterations across >200 disease-relevant cell types. Pharmaceutical partners including AstraZeneca, Merck, and Eli Lilly will use the information to evaluate medication targets, train sophisticated AI models, and investigate disease causes at a never-before-seen scale as the Atlas grows toward a 5 billion cell resource in the upcoming years. Such large?scale, high?resolution data significantly enhance the ability of AI systems to generate mechanistic insights and improve target discovery workflows.
The industry's general trend toward multi-omics and hyperscale datasets as the backbone of complex AI models, particularly those that need realistic cellular biology inputs for training, is reflected in this endeavor.
Breakthrough AI Models for Drug Development:[23,24]
Enchant, a cutting-edge AI platform from Iambic Therapeutics, uses multimodal transformer architectures to forecast the clinical and preclinical characteristics of drug candidates early in the development process. Enchant estimates important characteristics like pharmacokinetics (PK) and other drug qualities that are essential for clinical success by integrating vast amounts of preclinical and discovery stage data rather than depending just on the limited amount of clinical data. This methodology attempts to decrease development time, cost, and late stage failures by effectively bridging the gap between discovery data and downstream clinical outcomes. It has also established new predictive standards.
The ongoing development of Enchant (e.g., Enchant v2) shows that even with little clinical data, massive, multimodal AI models may perform better than earlier methods, representing a real step toward truly predictive, end-to-end AI platforms in drug discovery.
Broader Industry Momentum:[25]
AI is changing drug discovery, as evidenced by specific scientific projects, strategic alliances, and greater investment across biotech and pharma. As an illustration of the transition from experimental bottlenecks to AI-driven processes, large pharmaceutical companies are implementing AI tools to increase the effectiveness of clinical trials, lessen administrative costs, and improve data analysis.
All of these developments—big datasets like the Billion Cell Atlas, sophisticated multimodal models like Enchant, and wider industry adoption—showcase a real-world shift toward drug discovery ecosystems that are scalable, data-driven, and AI-enabled. In addition to speeding up scientific research, these changes raise the possibility of more potent, specialized, and individualized treatment options in the years to come.
CONCLUSION:
The field of medication development and discovery is changing as a result of artificial intelligence (AI) and machine learning (ML). According to this review, target identification, virtual screening, lead optimization, ADMET prediction, drug repurposing, and clinical trial design have all been greatly enhanced by AI-driven techniques, such as machine learning, deep learning, graph neural networks, natural language processing, and generative/reinforcement learning models. Artificial Intelligence (AI) has changed drug discovery from a primarily empirical process to a data-driven, predictive, and logical science by making it possible to analyze huge, complicated datasets and create innovative chemical structures.
Even with these revolutionary developments, a number of obstacles still exist. The interpretability of the model (the "black box" problem), data availability and quality, ethical and legal issues, and the requirement for a strong integration with experimental validation are some of the main drawbacks. Many of these obstacles should be removed by emerging solutions like explainable AI, hybrid AI-experimental frameworks, quantum computing, and foundation models, which will further improve the prediction accuracy, dependability, and scalability of AI systems.
In conclusion, artificial intelligence is now a major force behind pharmaceutical innovation rather than a supporting instrument. It represents a paradigm shift in the development of pharmaceuticals due to its capacity to expedite drug discovery, lower costs, increase clinical success rates, and enable personalized treatment. To fully fulfill AI's disruptive potential to revolutionize pharmaceutical R&D, interdisciplinary collaboration, large datasets, and ethical governance will be essential.
REFERENCES





Bhagwan Gite, Pankaj Shinde, Keshav Pawar, Omkar Fule, Mayur Sonar, Artificial Intelligence and Machine Learning in Drug Discovery: Trends, Techniques, Challenges, and Future Prospects., Int. J. of Pharm. Sci., 2026, Vol 4, Issue 2, 2661-2675. https://doi.org/10.5281/zenodo.18670978
 10.5281/zenodo.18670978
10.5281/zenodo.18670978
