
We use cookies to ensure our website works properly and to personalise your experience. Cookies policy
Swami Vivekanand College of Pharmacy, Udgir
Alzheimer’s disease (AD) is the most widespread neurodegenerative disorder and a leading cause of dementia seen in clinical practice. Although many therapeutic strategies have been explored, AD continues to present a major challenge for healthcare systems around the world. This article begins by summarizing the key biological processes that lead to neuronal damage in AD. It then critically examines important research on current pharmacological treatments and highlights recent innovations and developing therapies. The review covers the main characteristics, classification, FDA approval status, mechanisms of action, therapeutic benefits, and common adverse effects of both established and emerging drugs for AD management. Traditional drug classes discussed include cholinesterase inhibitors, monoclonal antibodies, and other therapies such as memantine, valproic acid, and rosiglitazone. The article also explores newer monoclonal antibodies—donanemab, gantenerumab, solanezumab, bapineuzumab, crenezumab, and semorinemab—and evaluates nutritional supplements like vitamin E (alpha-tocopherol) and caprylidene. Additional agents that target tau and amyloid pathways, such as methylthioninium (MT), leuco-methylthioninium bis (LMTM), and tramiprosate, are reviewed for their ability to inhibit A? aggregation. The discussion extends to antidiabetic and anti-inflammatory drugs recently proposed for AD therapy. These include NE3107, a compound with anti-inflammatory and insulin-sensitizing effects, and the commonly used antidiabetic drug metformin. Anti-neuroinflammatory agents— such as sodium oligomannate (GV-971), intravenous immunoglobulin infusions to reduce circulating A? components, and masitinib, a tyrosine kinase inhibitor affecting mast cells and microglia—are also highlighted.
Dementia has become a major worldwide public health concern. As outlined in the World Health Organization’s 2022 dementia research blueprint, approximately 55.2 million people are living with the condition globally. The prevalence in adults over 60 differs across regions—about 2.9% in Southeast Asia, 6.5% in Europe, and between 3.1% and 5.7% in other parts of the world. (1)
Unlike many other neurological disorders, Alzheimer’s disease is characterized by a striking buildup of misfolded proteins within the brain. These protein abnormalities accumulate to high levels, leading to the development of intracellular neurofibrillary tangles and extracellular amyloid plaques. (2)
Current explanations for the development of Alzheimer’s disease are largely informed by genetic research and neuropathological observations. These findings suggest that the disease arises from abnormal processing of two key proteins: amyloid precursor protein (APP) and tau. Evidence from molecular biology, genetics, and brain pathology supports the idea that age-related disruptions in the metabolism of APP and tau trigger a cascade of harmful events. This leads to the buildup of beta-amyloid (Aβ) fibrils and tau-based neurofibrillary tangles. The accumulation of these protein aggregates damages neurons and synapses in specific regions of the brain, ultimately resulting in cognitive decline.(3)
Genetic factors can play a direct role in the development of Alzheimer’s disease, particularly mutations in the APP, Presenilin 1, and Presenilin 2 (PS1/PS2) genes. These genes influence the production of amyloid-β peptides, and their abnormal activity contributes to the buildup of these peptides in the brain.
In addition to genetic causes, several modifiable and non-modifiable risk factors i10.1016/j.pneurobio.2010.01.002ncrease the likelihood of developing AD. (4)
Need of study
Alzheimer’s disease (AD) remains a significant global health issue, with its incidence increasing as the population ages and life expectancy rises. Although substantial research has been conducted, the disease’s underlying mechanisms are still not fully clarified, and existing therapies mainly provide temporary symptom control rather than slowing or preventing progression. The emotional, financial, and caregiving challenges faced by patients and families further emphasize the need for more comprehensive research. A deeper exploration of molecular mechanisms, early diagnostic indicators, and new therapeutic targets is vital for discovering treatments that can modify or halt the disease. Continued scientific investigation is therefore essential to enhance patient care, lessen the worldwide burden of AD, and support the development of more advanced and effective treatment options.
Aim and Objectives
To explore and evaluate the underlying mechanisms, diagnostic approaches, and current as well as emerging therapeutic strategies for Alzheimer’s disease in order to improve understanding and support the development of effective treatments.
What is Alzheimer Disease
Alzheimer’s disease (AD) is a form of dementia that is distinct from the normal memory decline seen with aging. It primarily affects older adults, especially those aged 65 and above. AD is a progressive, irreversible neurodegenerative disorder. Early manifestations often include trouble recalling recent events or conversations, along with apathy and depression. As the disease advances, individuals may experience increasing difficulty with communication, confusion, disorientation, poor decision-making, behavioral changes, and eventually problems with speaking, swallowing, and walking.
Multiple factors contribute to neurodegeneration in AD, including rare genetic mutations (accounting for less than 5% of cases), abnormal protein folding, neuronal membrane damage, mitochondrial dysfunction, oxidative stress, accumulation of toxic molecules, and ongoing neuroinflammation. One of the major obstacles in combating Alzheimer’s disease is the absence of reliable biomarkers that can enable early detection, effective prevention, and timely therapeutic intervention. (5,6)
Literature review
Alzheimer’s disease (AD) is a progressive neurodegenerative disorder and the leading cause of dementia worldwide, accounting for nearly 60–70% of cases. The literature describes AD as a multifactorial condition characterized by the accumulation of amyloid-β (Aβ) plaques, neurofibrillary tangles composed of hyperphosphorylated tau, oxidative stress, and chronic neuroinflammation. Early studies focused primarily on the amyloid cascade hypothesis, proposing that abnormal cleavage of amyloid precursor protein leads to Aβ aggregation and neuronal toxicity. Subsequent research expanded the understanding of AD pathogenesis by highlighting tau pathology, mitochondrial dysfunction, cholinergic neuron loss, and vascular contributions such as reduced cerebral perfusion and blood–brain barrier breakdown.
Clinical literature describes AD as a progressive condition beginning with mild memory loss and advancing to severe cognitive and functional impairment. Diagnostic advancements over the past decade include MRI for detecting hippocampal atrophy, PET imaging for amyloid and tau visualization, and cerebrospinal fluid biomarkers (Aβ42 decrease, tau increase). Recent breakthroughs include blood-based biomarkers such as plasma p-tau181 and p-tau217, which offer inexpensive and early detection.
Pharmacological literature identifies two major treatment categories: symptomatic therapies and disease-modifying approaches. Cholinesterase inhibitors (donepezil, rivastigmine, galantamine) and the NMDA receptor antagonist memantine remain standard symptomatic treatments. More recent research has focused on disease-modifying monoclonal antibodies like aducanumab, lecanemab, and donanemab, which aim to reduce amyloid burden, though their clinical benefits are modest and debated. Current studies also explore tau-targeting therapies, anti-inflammatory agents, gene editing, and stem-cell–based strategies.
Despite significant progress, the literature consistently emphasizes key limitations: the complexity of AD pathophysiology, late-stage diagnosis, difficulty crossing the blood–brain barrier, and high failure rates in clinical trials. However, future directions suggested by contemporary research include precision medicine, multimodal therapies targeting multiple pathological pathways, early screening using blood biomarkers, and lifestyle-based prevention strategies. Overall, existing literature reflects a shift from a single-pathway amyloid hypothesis to a more integrative view of AD as a multifaceted neurodegenerative disorder requiring early detection and combination therapies.
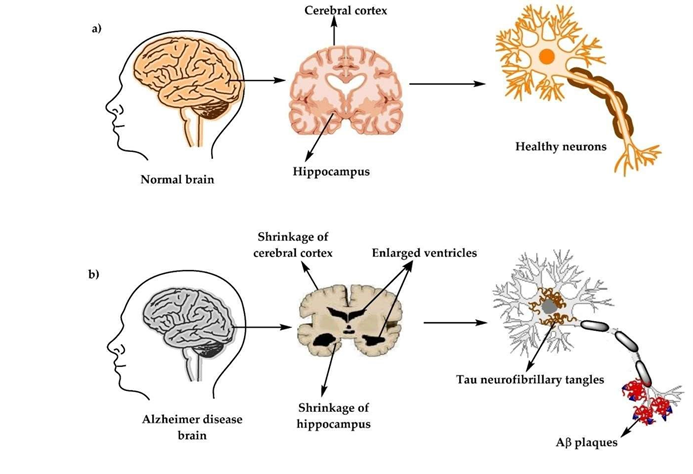
Fig 1 Alzheimer Disease
Historical background
Alois a German psychiatrist and neuropathologst, documented the first known case of Alzheimer’s disease in 1907. He had first encountered the patient, a 51-year-old woman named Auguste Deter, in 1901. Her husband, Karl, brought her to a mental institution after she began displaying unusual and disturbing behaviors, such as hiding belongings, accusing neighbors, and suspecting her husband of being unfaithful. She also gradually lost the ability to manage everyday household tasks like cooking and cleaning.
Under Alzheimer’s care in a Frankfurt mental hospital, her symptoms were carefully observed. Although she was able to speak, she struggled to write even her own name. She could identify simple objects, such as a pencil, yet could not name the food on her plate. Her behavior fluctuated—she could be calm and courteous at times, but loud, agitated, or inappropriate at others. Based on her symptoms, Alzheimer diagnosed her condition as “presenile dementia,” a term he used long before the disorder came to bear his name. (7,8)
Classification of Alzheimer Disease
Cause of Alzheimer Disease
Research into what causes Alzheimer’s disease (AD) has been ongoing for decades, and many theories have been suggested to explain its origin (see Box 1). Yet none of these proposed mechanisms has fully explained the disease, and several have been discarded over time. Today, scientists continue to investigate multiple biological processes that may drive neurodegeneration, such as chronic inflammation, oxidative damage, metabolic and lipid imbalance, dysfunction of the neurovascular unit, and abnormalities in protein folding and spread. Among these, amyloid-β accumulation and tau hyperphosphorylation remain major areas of focus.
Despite this, the true initiating cause of AD remains unknown. It has become increasingly clear that a single explanation is unlikely to fully capture the complexity of the disease. The history of the amyloid-β hypothesis illustrates this well. Although amyloid-β was originally viewed simply as a diagnostic marker (even by Alzheimer and Fischer themselves), it was later proposed as a direct cause of the disease in the landmark “amyloid cascade hypothesis” by Hardy and Higgins. This hypothesis suggested that amyloid-β plays an active role in triggering neurodegeneration, but it was not originally supported by solid AD-specific evidence.
While amyloid-β buildup is required for an AD diagnosis based on current guidelines, there is only limited proof that it actually causes the disease. Moreover, numerous clinical trial failures involving anti-amyloid therapies have cast further doubt on the hypothesis—even in familial AD, where the theory should theoretically fit best. Additional challenges arise from findings of individuals with genetic mutations (such as protective APOE3 or reelin variants) who still develop amyloid-β deposits but show reduced tau pathology and milder symptoms.
These observations suggest that in a subset of people, amyloid-β changes might indeed act as the first step in the disease process. However, it is unrealistic to assume that this mechanism applies universally to all individuals with AD, given the disease’s highly complex and multifactorial nature.
Pathology of Alzheimer Disease
The exact disease mechanism of Alzheimer’s disease is still not fully understood. However, two major pathological features are consistently observed. The first is the buildup of amyloid-β (Aβ) fragments, which form plaques around the outside of neurons. The second is the presence of abnormal, twisted tau protein strands—known as neurofibrillary tangles—inside the neurons. Over the years, multiple theories have been proposed to explain how these abnormalities contribute to the development and progression of AD, as outlined below.(9)
Amyloid precursor Protein
When amyloid precursor protein (APP) is abnormally cut by the enzymes β-secretase and γsecretase, it leads to the formation and buildup of Aβ40 and Aβ42 peptides outside neurons and outside blood vessels. (10,11)
The amyloid cascade hypothesis, one of the most widely discussed theories of Alzheimer’s disease (though still debated), proposes that the accumulation of Aβ peptides in the brain—caused by an imbalance between their production and clearance—is the primary event driving the disease. Other pathological changes, such as the formation of neurofibrillary tangles, are thought to occur as a consequence of this initial Aβ buildup. (12)
The Aβ peptide, consisting of 36 to 43 amino acids, is generated through enzymatic cleavage of the amyloid precursor protein (APP), a naturally occurring protein that is essential for maintaining normal brain function. (13,14)
The APP gene is found on chromosome 21, which helps explain why early-onset Alzheimer’s disease is more common in people with trisomy 21 (Down syndrome) and in rare cases of familial early-onset AD caused by duplication of the APP gene. Overexpression of APP is thought to increase the production of Aβ peptides in the brain, leading to their accumulation and deposition.(15)
A clear understanding of the various biological pathways involved in Alzheimer’s disease (AD) is essential for designing effective therapies. However, the exact sequence of events that drives AD development is still not fully understood. Current evidence suggests that the build-up of insoluble beta-amyloid plaques and tau tangles disrupts normal neuronal activity and communication in the brain.
Beta-amyloid plaques form when amyloid precursor protein (APP) is processed through the amyloidogenic pathway, producing sticky beta-amyloid fragments that cluster together. These aggregates accumulate both inside and around neurons, interfering with synaptic signaling and triggering harmful processes such as inflammation and oxidative stress, which ultimately damage brain cells. (15)
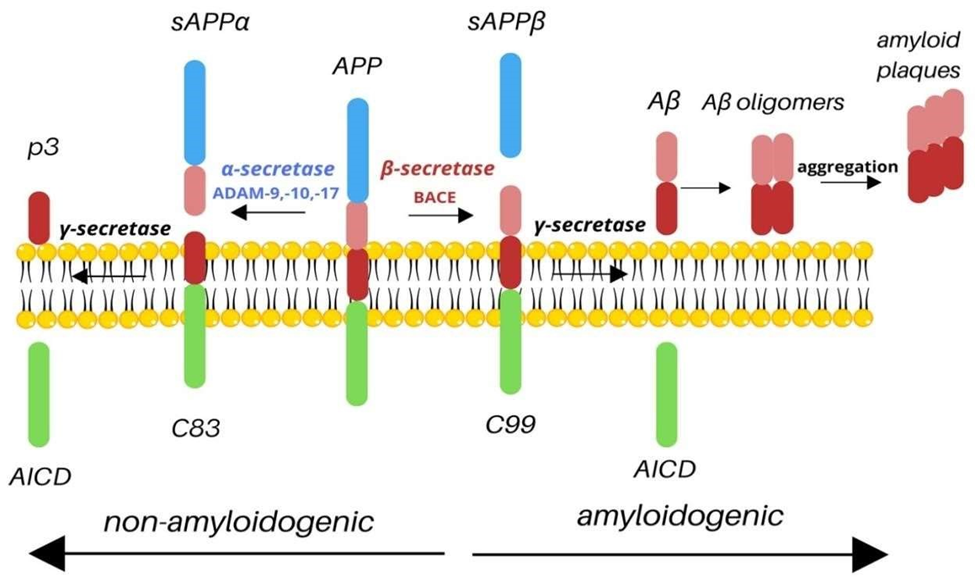
Fig 2 : Amyloid precursor Protein
Tau Proteins
Tau is a microtubule-associated protein encoded by the MAPT gene on chromosome 17 (17q21). Alternative splicing of this gene generates six different tau isoforms.(16)
Tau protein primarily helps promote tubulin polymerization, stabilize microtubules, and facilitate the transport of intracellular organelles along microtubules. When tau becomes hyperphosphorylated, it loses these functions, disrupting microtubule structure, causing neuronal damage, and contributing to cytotoxicity.(17)
Tau protein is a microtubule-associated protein that is essential for axonal growth and neuronal development by ensuring the stability of microtubule structures. (18)
Studying the function of tau protein and its pathological changes in Alzheimer’s disease is essential for creating therapies that target tau-related neurotoxicity and slow disease progression. In healthy neurons, kinases and phosphatases maintain a balance between phosphorylation and phosphorylation of tau, controlling the addition or removal of phosphate groups and supporting microtubule stability. (19 , 20 )
Tau is a type of microtubule-associated protein that helps maintain the stability and structure of microtubules. These microtubules are essential for the growth, differentiation, and proper connectivity of neurons. The human TAU gene is located on chromosome 17.
In Alzheimer’s disease and in certain inherited mutations affecting the TAU gene, tau proteins become excessively phosphorylated by several protein kinases such as MAPKs, CDK5, and GSK3β. This abnormal hyperphosphorylation weakens tau’s ability to bind and stabilize microtubules. As a result, microtubules break down, disrupting the internal transport system of neurons and impairing communication between nerve cells.
The overly modified tau proteins then start accumulating inside neurons. These hyperphosphorylated tau molecules assemble into paired helical filaments, which eventually form neurofibrillary tangles (NFTs)—a key pathological hallmark of Alzheimer’s disease.
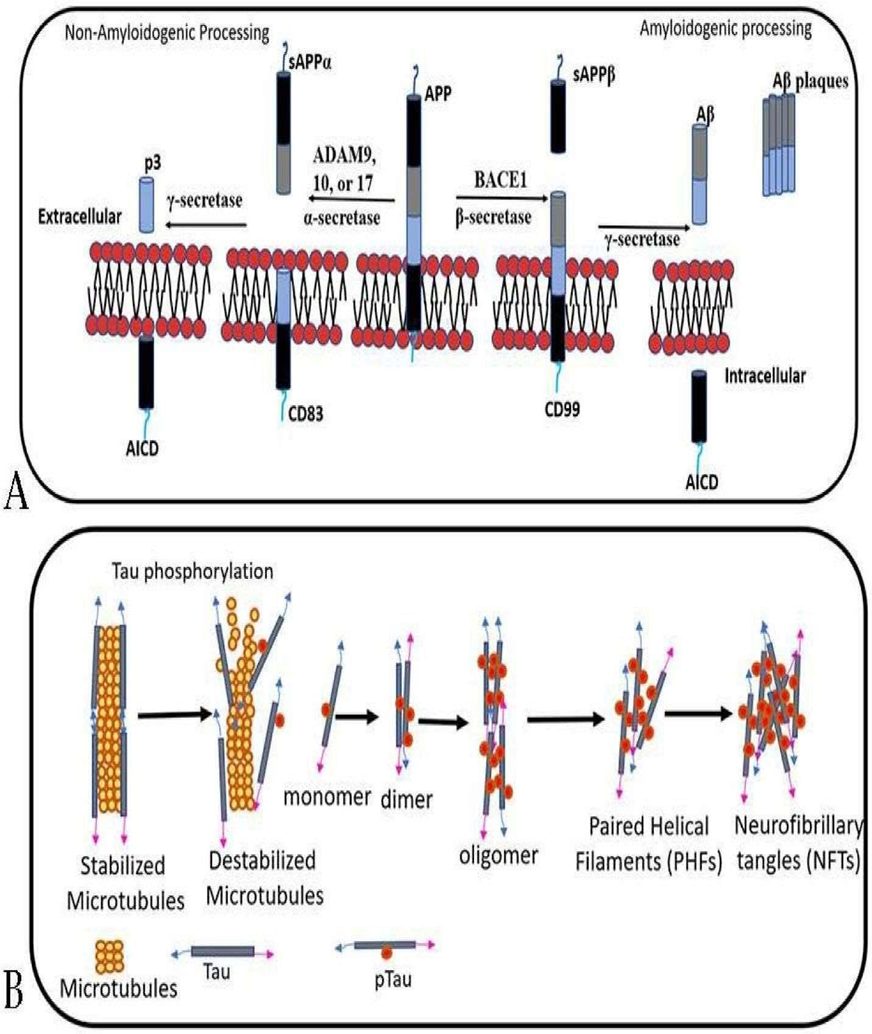
Fig 3 : Tau Proteins
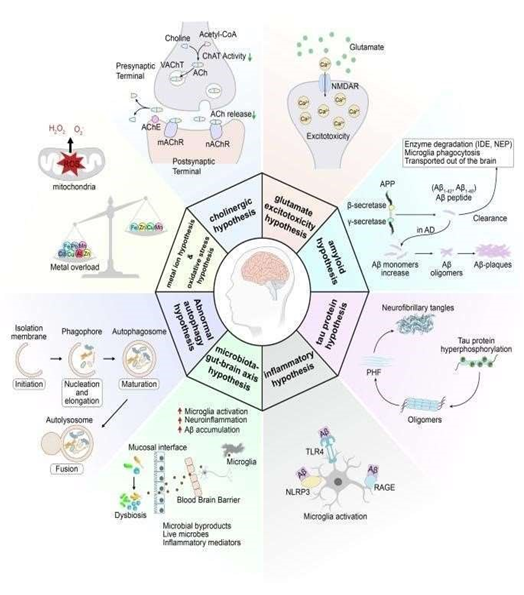
Fig 4 : Mechanism
Mechanisms of Alzheimer Disease
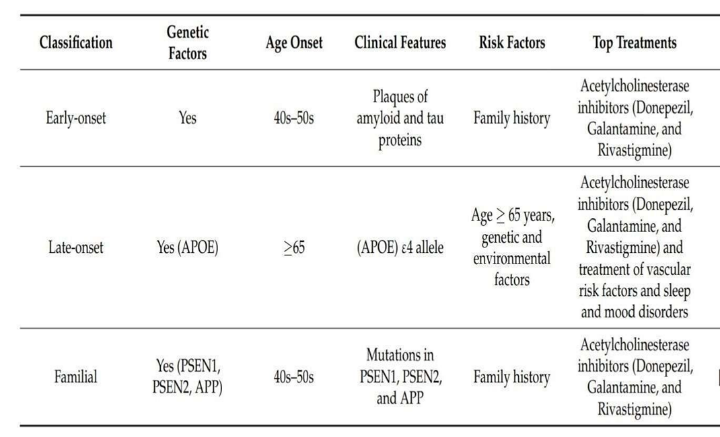
Many hypotheses have been suggested to explain the development of Alzheimer’s disease, but a single, comprehensive theory has not yet been established, likely because of the disease’s complexity. AD is generally classified into two main types: familial, which represents 1–5% of cases, and sporadic, which accounts for over 95% of cases. (21)
Familial Alzheimer’s disease (FAD) is mainly caused by autosomal dominant mutations in the APP, presenilin 1 (PS1), and presenilin 2 (PS2) genes. It typically appears between the ages of 30 and 65 and progresses rapidly. In contrast, sporadic or late-onset Alzheimer’s disease (SAD) generally occurs after age 65 and is influenced by a mix of genetic predisposition, environmental factors, and other health conditions. (22 ,23 )
Genome-wide association studies (GWAS) and large-scale meta-analyses have uncovered multiple genetic risk loci linked to sporadic Alzheimer’s disease (SAD), highlighting pathways involved in immune function, lipid metabolism, amyloid-β plaque formation, neurofibrillary tangles, and endocytosis. However, many genetic loci contributing to the disease are still unknown. (24,25 )
Along with neuronal damage, Alzheimer’s disease (AD) is also marked by a noticeable increase in the number of senile plaques. According to the cholinergic hypothesis, there is a strong link between the loss of cholinergic neurons in the basal forebrain and the cognitive decline seen in AD. These neurons are essential parts of the brain’s cholinergic network and play a major role in memory, attention, and overall cognitive processing.
Most cholinergic neuron cell bodies are located in regions such as the medial septal nucleus (MSN), the diagonal band of Broca (DBB), the nucleus basalis of Meynert (NBM), and the substantia innominata (SI). Research shows that neurons in the NBM are especially vulnerable to degeneration in AD, likely due to reduced support from nerve growth factor (NGF).
Acetylcholine (ACh) is produced in neurons from choline and acetyl-CoA by the enzyme choline acetyltransferase (ChAT). It is then packed into synaptic vesicles through the vesicular acetylcholine transporter (VAChT). Once a nerve impulse arrives, ACh is released into the synaptic cleft, where it activates muscarinic (mAChR) and nicotinic (nAChR) receptors on the postsynaptic neuron. After signal transmission, acetylcholinesterase (AChE) breaks down ACh into choline, which is taken back up by the presynaptic neuron for reuse.
In AD, reduced ChAT activity—combined with the negative impact of amyloid-β on ACh production, release, and metabolism—results in lower acetylcholine levels. This decline interferes with key functions such as learning, memory, movement regulation, and sleep patterns.
The cholinergic hypothesis has played a major role in early AD research and drug development. AChE inhibitors (AChEIs) like donepezil, rivastigmine, and galantamine, introduced over 20 years ago, are still central to AD therapy.
Risk factors
1. Genetic factors
Alzheimer’s disease can also be grouped based on the age at which symptoms first appear. Early onset AD occurs before the age of 65 and represents roughly 4–6% of all cases. Late-onset AD, the more common form, begins at or after age 65. In addition to differences in onset age, these two forms show variations in their clinical features, cognitive patterns, brain pathology, and neuroimaging findings.(26)
Over 30 dominant mutations have been identified in the APP gene (on chromosome 21q21), accounting for roughly 15% of early-onset autosomal dominant Alzheimer’s cases. PSEN1 mutations (on chromosome 14q24.3) are the most common, responsible for about 80% of earlyonset AD cases, while PSEN2 mutations (on chromosome 1q31–q42) contribute to around 5% of cases.(27)
2. Hypertension
Several acquired or lifestyle-related factors can raise the likelihood of developing Alzheimer’s disease. The most frequently reported among these are cerebrovascular disorders, along with diabetes, high blood pressure, obesity, and abnormal lipid levels.(28)
3. Acquired risk factor
The links between these risk factors and the development of Alzheimer’s disease will be explored in the next sections.
Additionally, protective factors—such as greater cognitive reserve, regular physical exercise, and healthy dietary habits, as highlighted by Mayeux & Stern (2012)—will also be discussed for their role in lowering disease risk. (29)
3. Obesity
The contribution of obesity to Alzheimer’s disease risk remains unclear, as research findings are highly variable. However, a meta-analysis conducted by Profenno, Porsteinsson, and Faraone (2010) suggests… (30)
Research shows that obesity (BMI ≥ 30 kg/m²) is strongly and independently linked to a higher risk of developing Alzheimer’s disease. However, findings vary across studies. For example, a meta-analysis by Fitzpatrick et al. (2009) found that obesity during midlife increases the likelihood of developing dementia (HR: 1.39; 95% CI: 1.03–1.87). Interestingly, the same study reported that in older adults, obesity actually appears to be associated with a lower risk of dementia (HR: 0.63; 95% CI: 0.44–0.91). They also noted that being underweight (BMI < 20 kg/m²) raises dementia risk as well (HR: 1.62; 95% CI: 1.02–2.64). In later life, unintentional weight loss often occurs alongside other health problems, reflects declining health, and may even begin up to 10 years before dementia symptoms appear. (31)
4. Diet
The makeup of phospholipids is crucial for proper neuronal membrane function. Therefore, sufficient consumption of nutrients like DHA, EPA, uridine monophosphate, choline, folate, vitamins B6, B12, C, and E, as well as selenium, supports healthy phospholipid production. This, in turn, helps maintain synaptic activity and provides protection against neurodegeneration. Antioxidants help protect cells by blocking the harmful effects of reactive oxygen species and also support membrane stability. DHA assists in the removal of Aβ peptides, and along with choline and uridine, contributes to the formation and maintenance of neuronal membranes. (32)
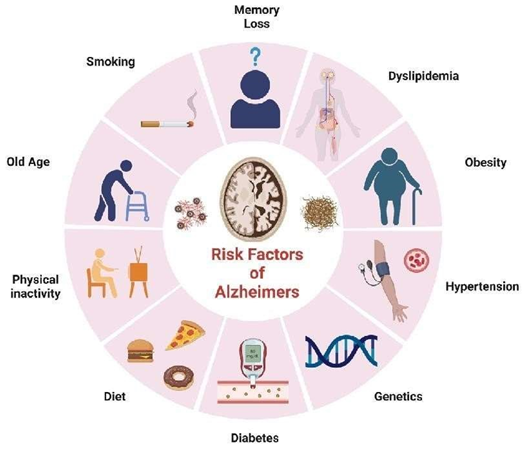
Fig 5: Risk Factors
Diagnosis of Alzehmer Disease
AD is considered a clinicopathological disorder, meaning that a definite diagnosis requires two key elements:
Treatment of Alzheimers Disease
Behavioral and psychiatric symptoms of dementia (BPSD)—also known as neuropsychiatric symptoms—are very common in Alzheimer’s disease. These include agitation, depression, apathy, psychosis, anxiety, aggression, disinhibition, and sleep-related problems. Such symptoms significantly reduce patients’ quality of life and place a heavy emotional and physical burden on caregivers. Drug therapy for BPSD is often unpredictable, and many clinical studies have shown limited effectiveness. This section highlights the management of agitation, depression, apathy, psychosis, and sleep disturbances in Alzheimer’s disease.
Agitation is frequently observed in AD and presents as restlessness, aggression, and irritability. Both typical antipsychotics (e.g., haloperidol) and atypical antipsychotics (e.g., risperidone, quetiapine) may be used but carry significant risks, including sedation and higher mortality. In 2023, the FDA approved brexpiprazole for agitation associated with AD, demonstrating moderate benefit at 2 mg/day. Other options such as dextromethorphan/quinidine and escitalopram may help, though more research is needed to confirm their effectiveness. Non-drug approaches—such as music therapy, structured physical activities, and environmental modifications—are preferred as first-line treatments.
Depression commonly accompanies AD and is characterized by low mood, reduced interest, and social withdrawal. SSRIs like citalopram and sertraline provide modest improvement, while mirtazapine may help both depressive symptoms and sleep. Psychological therapies such as cognitive behavioral therapy (CBT) and mindfulness can be useful additions to treatment.
Apathy, which involves reduced motivation and emotional responsiveness, is another frequent symptom. Evidence shows that methylphenidate can improve apathy over a short term (around 3 months) with minimal side effects. Nonpharmacological approaches—such as behavioral activation and structured daily routines—may also help in managing apathy.
Psychosis, including delusions and hallucinations, tends to appear in the later stages of AD. Antipsychotic medications may be prescribed when symptoms are severe, but they carry risks including cardiovascular problems. Non-drug strategies such as cognitive therapy and environmental adjustments can help reduce psychotic symptoms safely.
Sleep problems are common and tend to worsen as AD progresses, further reducing clearance of amyloid-beta from the brain. Melatonin has shown mixed results, with some studies reporting benefits and others showing no significant effect. Suvorexant, approved in 2020 for insomnia in mild to moderate AD, has been shown to improve sleep duration and reduce nighttime awakenings. However, because suvorexant is a Schedule IV controlled substance in the U.S., its use remains limited. Nonpharmacological methods—including CBT for insomnia (CBT-I) and bright light therapy—are effective for improving sleep. Managing other BPSD symptoms such as anxiety, depression, and agitation can also enhance sleep quality.
Future prospects
Observation of Alzheimers Dieases
Alzheimer’s disease (AD) is a chronic, progressive neurodegenerative disorder that predominantly affects older adults and is clinically characterized by a gradual decline in cognitive, behavioral, and functional abilities. Observations in patients with AD consistently reveal a pattern of memory loss, impaired reasoning, and progressive deterioration in daily activities, reflecting underlying pathological changes within the brain. The earliest and most prominent clinical observation is episodic memory impairment, especially difficulty in retaining newly learned information. As the disease advances, additional cognitive domains—including language, visuospatial skills, judgment, and executive functioning— become significantly affected.
Neurological examinations and neuroimaging studies provide further observations supporting the diagnosis of AD. Structural MRI typically shows cortical atrophy, most markedly in the hippocampus, entorhinal cortex, and medial temporal lobes—areas associated with learning and memory. Functional imaging (such as FDG-PET) often demonstrates hypometabolism in the posterior cingulate cortex, precuneus, and parietotemporal regions. Amyloid PET imaging reveals extracellular deposition of amyloid-β plaques, whereas tau PET highlights the presence of neurofibrillary tangles, which correlate strongly with disease severity. These pathological hallmarks are mirrored in cerebrospinal fluid (CSF) findings, where reduced Aβ42 levels and elevated total tau and phosphorylated tau serve as key biomarkers.
Behavioral and psychological symptoms are also commonly observed and may appear at any stage of the disease. These include apathy, irritability, agitation, depression, sleep disturbances, and, in later stages, profound confusion and wandering tendencies. As neurodegeneration spreads to additional cortical and subcortical regions, patients experience progressive loss of independence, ultimately requiring full-time caregiving and assistance with basic activities such as eating, bathing, and mobility.
Postmortem histopathological observations confirm the widespread presence of amyloid-β plaques, neurofibrillary tangles composed of hyperphosphorylated tau, synaptic loss, gliosis, and neuronal death, particularly in the hippocampus and association cortices. These findings correlate with the severity of cognitive impairment observed during life and reinforce the dual clinicopathological nature of the disease.
CONCLUSION
Alzheimer’s disease is a complex neurodegenerative condition characterized by progressive cognitive decline and significant morbidity. While the amyloid and tau hypotheses continue to guide much of the therapeutic development, research in the past decade has highlighted the pivotal roles of neuroinflammation, mitochondrial dysfunction, oxidative stress, genetics, and vascular impairment. Current treatments—including cholinesterase inhibitors and NMDA receptor antagonists—offer only modest symptomatic benefit, and recently approved monoclonal antibodies provide limited yet important steps toward disease modification. However, challenges such as late diagnosis, heterogeneity among patients, and the difficulty of delivering drugs across the blood–brain barrier hinder therapeutic success. Moving forward, the future of AD management will rely on early screening tools, blood-based biomarkers, precision medicine approaches, and multimodal therapies targeting multiple pathological pathways. Although no cure currently exists, advances in technology, biomarker science, and neurobiology provide cautious optimism that more effective treatments may emerge.
REFERENCES

Bhakti More, Navnath Bendke, Pratiksha Swami, Sharmin Shaikh, Dr. Ganesh Talsarwad, Comprehensive Review on Alzheimer Disease: Causes, Risk Factors, Pathology, Mechanisms, Int. J. of Pharm. Sci., 2026, Vol 4, Issue 2, 942-957. https://doi.org/10.5281/zenodo.18509183
 10.5281/zenodo.18509183
10.5281/zenodo.18509183