
We use cookies to ensure our website works properly and to personalise your experience. Cookies policy
JKK Munirajah Institute of Health Sciences College of Pharmacy, T.N Palayam, Ero
The autoimmune condition known as myasthenia gravis (MG) affects the neuromuscular junction. Autoantibodies against the acetylcholine receptor (AChR) are present in about 74–88% of gMG patients. A variable percentage of the remaining 20% is composed of antibodies to muscle-specific kinase [MuSK; MuSK antibody positive myasthenia gravis (MuSK-MG)]. Ocular, bulbar, respiratory, axial, and limb muscles all show characteristic fatigable weakness due to the production of antibodies against the acetylcholine receptor (AChR) in this classic autoimmune disease. Thymus gland abnormalities, stress, infections, operations, and adverse drug reactions can also bring it on. When a person has this illness, their thymus grows larger than usual; this condition is also known as thymic hyperplasia. This study concentrates on the immunological mechanisms in MG, the new drugs that target them, and the safety and effectiveness of the clinical data. These include B-cell depletion drugs (anti-CD 19 and 20 and B-cell activation factor [BAFF] inhibitors), complement C5 inhibitors, Fc receptor inhibitors, and subcutaneous immunoglobulin (SCIG).
The classic autoimmune disease associated with certain autoantibodies at the neuromuscular junction is myasthenia gravis (MG). Corticosteroids, azathioprine, that have been successful in managing myasthenia [8]. Globally, the illness is thought to affect 70–300 people per million [10]. The pathophysiology and immunological regulation of MG depend on B-cell and T-cell activation [5]. While many patients can benefit from conventional therapy choices such as symptomatic therapies and systemic immunosuppression, not all individuals can [13]. The first commonly used immunosuppressive treatment for MG was corticosteroid therapy. High-dose prednisone (100 mg/d or every other day) was used in the initial reports of a positive response in MG. Prednisone had a significant effect on myasthenic patients in early clinical trials; at least 80% of them demonstrated either medical remission or noticeable improvement [7]. Oral corticosteroids are first used to create remission, then either non-steroidal immunosuppressants or low doses of oral corticosteroids are used to maintain the condition over the long term [9]. The Both chronic symptoms and acute exacerbations, such as myasthenic crises, which are life-threatening events requiring hospitalization and typically critical care support due to bulbar and respiratory muscle failure, are treated medically. One of the disease's most distinctive features is symptom fluctuation, which is typically linked to various triggers including infections, vaccinations, surgery, or other stressful circumstances [6]. Human leukocyte antigen (HLA) DR3-D8 and thymic follicular hyperplasia are strongly associated with early onset MG, which is more common in women. On the other hand, despite having an atrophic thymus, late-onset MG frequently affects men, has no HLA link, and may exhibit anti-striational antibodies [9].
Mechanisms of action of steroids and other immunosuppressants, as well as randomized and nonrandomized data supporting their effectiveness in treating myasthenia gravis, was examined in this review.
1)PATHOLOGY OF MYASTHENIA GRAVIS:
Although environmental causes, including viral infections and genetic vulnerability, have been suggested, the exact cause of the autoimmunity in MG has not been determined. The pathophysiology of AChR-MG is thought to be significantly influenced by pathological alterations in the thymus. The most widely recognized theory focuses on the intrathymic failure of self-tolerance in AChR-MG [9]. Clinically, individuals are classified as having either generalized MG, which involves weakness of any voluntary muscle, or ocular myasthenia, which is defined by complaints of ptosis, diplopia, or both. From extremely limited symptoms, especially in the bulbar muscles, to broad muscle weakness, such as respiratory insufficiency leading to respiratory failure, generalized weakness can take many forms [2]. Immune intolerance in acetylcholine receptor antibody-positive myasthenia gravis (AChR+MG) is thought to be largely caused by thymus dysfunction. Antibodies against AChR are primarily immunoglobulin (Ig) G1 and IgG3. In addition to activating the complement cascade to form the membrane attack complex that eliminates postsynaptic membrane folds, these autoantibodies also crosslink the receptors at the neuromuscular junction, preventing acetylcholine from binding to AChR and causing their internalization and degradation. As a result, transmission across the neuromuscular junction is unable to provide consistent muscle activation, leading to observable weakness [10]. Concordance is present in about 35% of monozygotic MG twins, indicating that environmental factors are the primary cause of the disease. However, Mendelian inheritance does not apply to MG. The likelihood of MG in family members is roughly 1000 times higher than in the general population [3].
2)TREATMENT:
Most MG sufferers are able to lead nearly normal lives thanks to the current treatment options. Additionally, in patients who are properly treated with long-term immunosuppression and under the supervision of qualified professionals, rapid worsening with respiratory failure (myasthenic crisis) is very uncommon (less than 2%).
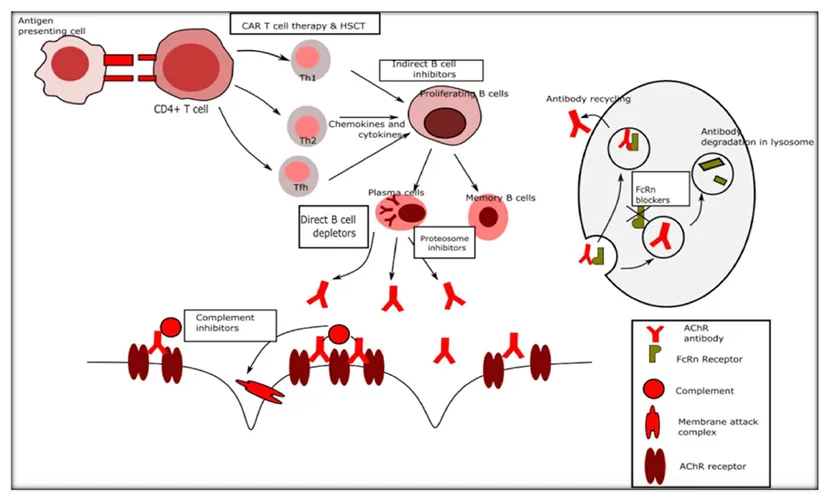
FIGURE1| Immune targets for novel therapies in myasthenia gravis.
Despite the possibility of related side effects, many patients need to take immunosuppressive medicine for years or even forever [22]. Immunosuppressive drugs work by either activating or suppressing target genes, which results in a variety of changes, such as reducing circulating T cells and suppressing antigen production (corticosteroids), interfering with T and B cell proliferation by cell cycle arrest (azathioprine, methotrexate, and mycophenolate), inhibiting T cell activation (cyclosporine, tacrolimus), inhibiting antigen-presenting cell interaction with T cells and Fc receptor blockade, among other actions (IVIG) [9]. The immune system and possible targets for new treatments are shown in Figure 1.
Three distinct mechanisms-classical, lectin, and alternative pathways—activate a group of proteins that mediate it. Through the conventional pathway, the autoantibodies in MG (especially IgG1 and IgG3 AChR antibodies, but typically not IgG4 MuSK antibodies) trigger the complement system [9]. The cascades of the classical and common complement pathways are initiated after IgG binds to the AChR epitopes. As a result of the cascade's latter stages, C5 convertase is formed, dividing C5 into C5a and C5b. The membrane attack complex (MAC), which integrates into the cell membrane and causes cell damage and lysis, is formed when C5b joins forces with C6–C9 factors [8]. A humanized recombinant monoclonal antibody called eculizumab binds to the C5 fragment and stops it from being cleaved, forming the complement terminal complex (MAC), and causing subsequent damage to the NMJ [18].
A recombinant humanized monoclonal antibody called eculizumab attaches itself to the C5 complement protein and prevents it from cleaving and forming the C5b-9 membrane attack complex. Following successful trials, it was recently licensed in late 2017 for the treatment of adult patients with generalized MG who are positive for the AChR antibody [7]. Although the response rate was not 100%, and most patients needed to continue receiving chronic therapy with different ISTs, a large phase 3 trial (REGAIN) showed significant advantages in individuals with resistant generalized MG. For 26 weeks, 125 patients with refractory generalized MG were randomized to receive intravenous eculizumab or an additional medicine to their current IST therapies, except for PLEX and IVIG. Induction dosage was 900 mg on days 0, 7, 14, and 21; maintenance dosage was 1,200 mg every other week for 26 weeks. The change from baseline to week 26 in the total score of MG Activities of Daily Living (MG-ADL), as determined by worst rank ANCOVA, was the primary effectiveness objective. Several secondary end-point measurements, including change in MG-ADL, Quantitative MG (QMG) score, and MG-Quality of Life (MG-QOL15), indicated significant improvement from baseline values in the eculizumab group when compared with placebo, but the trial did not satisfy the primary outcome efficacy criteria [8]. Meningococcal immunization is required beginning eculizumab therapy. Complement inhibition has the potential to significantly alter the way we treat MG patients [7].
2) ZILUCOPLAN:
Zilucoplan is a synthetic macrolide peptide inhibitor that functions similarly to eculizumab by blocking the cleavage of C5 complement protein into active C5a and C5b fragments, hence preventing the subsequent generation of MAC [8]. 44 patients with AChR Ab+ gMG participated in a phase 2, randomized, double-blind, placebo-controlled research to assess the clinical impact of subcutaneous (SC) zilucoplan. When compared to the placebo group, patients receiving zilucoplan 0.3 mg/kg daily for 12 weeks showed a statistically significant improvement in both primary and secondary endpoints. Zilucoplan has a good safety record with few adverse effects and no meningococcal infection. Zilucoplan's safety and effectiveness in gMG are being confirmed by a phase 3 trial [18].
3) RAVULIZUMAB:
Eculizumab's Fc region has amino acid changes that give it a high affinity for C5 and an instantaneous and long-lasting decrease in C5. Ravulizumab, another humanized monoclonal antibody, is a new C5 complement inhibitor [8]. FDA-approved ravulizumab for the treatment of PNH and aHUS following clinical trials that showed it to be just as effective as eculizumab, albeit with a lower dosage schedule (every 8 weeks as opposed to every 2 weeks). There is a phase 3 clinical trial in progress for refractory MG. Zilucoplan is a little peptide that prevents C5 from being cleaved. 44 patients with AChR Ab+ gMG participated in a phase 2, randomized, double-blind, placebo-controlled research to assess the clinical impact of subcutaneous (SC) zilucoplan. When compared to the placebo group, patients who received zilucoplan 0.3 mg/kg daily for 12 weeks showed a statistically significant improvement in both primary and secondary endpoints [18].
IgG plasma levels are decreased by FcRn inhibitors, which also increase their clearance and prevent recycling. A possible treatment strategy for AChR Ab+ and MuSKAb+MG may be FcRn inhibition [18]. The neonatal Fc receptor (FcRn), which is essential for IgG antibody recycling, is expressed on the surfaces of endothelial cells. Circulating antibodies attach to FcRn and internalize. Eventually entering lysosomes, but are normally recycled back into circulation. Antibodies in general, including the subgroup of antibodies that cause disease, are removed by proteolysis when FcRn inhibitors interfere with this binding within the lysosome. Within days of the first therapy, this leads to a notable decrease in the number of circulating antibodies [2].
1)EFGARTIGIMOD:
Endothelial cells are among the many cells and tissues that express the neonatal fragment crystallizable (Fc) receptor (FcRn), which contributes to the recycling of IgG and lengthens its half-life in the bloodstream by reducing its degradation in the Lysosomes. As a result, several FcRn blockers are at various phases of research for MG and other antibody-mediated disorders; in 2021, the FDA approved Efgartigimod for AChR Ab+ MG. The Fc component of IgG1, which has been modified to improve its affinity for the IgG binding site of FcRn, makes up efgartigimod [4]. 24 AChR Ab+ gMG patients participated in a phase 2 randomized 1:1 trial. The efgartigimod group experienced a quick and prolonged drop in IgG (70.7%) and AChR Ab blood levels (40–70%) as compared to the placebo group. Quantitative myasthenia gravis (QMG), myasthenia gravis activities of daily living profile (MG ADL), myasthenia gravis composite (MGC), and myasthenia gravis quality of life 15-item (MG-QoL15r) scores all showed a notable clinical improvement at the same time. The tolerance to efgartigimod was good. Headache and decreased monocyte count were the most common treatment-emergent adverse events (TEAEs). There is presently a phase 3 clinical trial in progress [18].
2)ROZANOLIXIZUMAB:
Rozanolixizumab is a humanized monoclonal antibody with a high affinity. IgG4P Ab takes direct aim at FcRn for abusing SC administration. 43 severe AChRAb+ and MuSKAb+gMG patients were randomly assigned to receive SC 7 mg/kg rozanolixizumab or placebo in a phase 2, placebo-controlled study. After 4 weeks, they were re-randomized to receive 3 weekly doses of either 4 or 7 mg/kg [18]. To assess safety, healthy participants were randomly assigned to receive a single intravenous or subcutaneous infusion of razonolixizumab at dosages of 1, 4, or 7 mg/kg in a phase I randomized placebo-controlled study. Headache (38.9%), vomiting (25%), nausea (19.4%), and pyrexia (19.4%) were the most frequent side effects, and they all happened more often with intravenous administration than with subcutaneous treatment. After peaking at 7–10 days, the decrease in IgG concentration progressively reverted to baseline every day [8].
C) B CELL TARGETING AGENTS:
One significant area of therapeutic exploration for MG is B-cell targeting. While a phase III trial employing rituximab within a year of disease onset improved clinical status, a phase II study of rituximab, a chimeric antibody directed at targeting CD20 on B-cells, in treatment-resistant MG failed to accomplish its primary objective [2].
i)RITUXIMAB:
A chimeric monoclonal antibody called rituximab targets the B-cell surface marker CD20. It effectively lowers circulating B-cell numbers and may be useful in treating autoimmune disorders caused by antibodies due to its ability to target autoreactive B-cell clones. Numerous case studies have indicated benefits for patients with MUSK MG and refractory MG [21]. RTX is a murine-human chimeric anti-CD20 glycoprotein monoclonal antibody that was created in the 2000s to treat cancer and other autoimmune diseases. The transmembrane protein CD20 is expressed by B cells but not by plasmablasts or long-lived plasma cells. It can destroy CD20+ cells through a variety of methods. Complement-mediated cytotoxicity and antibody-dependent cell-mediated cytotoxicity are among the direct effects of RTX, whereas apoptosis, structural alterations, and chemotherapy-sensitization of cancer cells are among the indirect effects. Additionally, RTX boosts Treg cells, which benefits MG immunology [8]. According to a recent comprehensive evaluation of the available retrospective rituximab studies, 72% of MuSK patients, 30% of AChR antibody patients, and 44% of both groups together achieved the Modified MFGA postintervention scale of minimum manifestation status or better [7].
i)BELIMUMAB:
Belimumab, an approved treatment for SLE, is a human IgG1λ monoclonal antibody that blocks B lymphocyte stimulator (BLys) against BAFF [7]. BAFF is a costimulator for B-cell survival and function and a member of the tumor necrosis factor (TNF) superfamily. The binding of BAFF to the B cell receptor keeps the autoantibody-producing B cells alive by preventing their death. Transgenic mice overexpressing BAFF have an excess of mature B cells, autoantibodies, and an overall increased autoimmune response, whereas animals lacking BAFF exhibit a marked drop in B cells and hypogammaglobulinemia [8]. There was no statistically significant difference between the belimumab and placebo groups in the key efficacy endpoint (QMG score) at 24 weeks in the phase 2 randomized, placebo-controlled clinical trial. The most common TEAEs in the belimumab (78%) and placebo (91%) groups were headache, diarrhoea, back pain, illness, and nausea [8].
ii) BORTEZOMIB:
Bortezomib is a dipeptide that causes misfolded or unfolded proteins to accumulate in plasma cells, ultimately leading to cell death, by blocking proteasome function. The medication boratezomib, which is used to treat multiple myeloma and "mantle cell lymphoma," seems to be a new treatment for autoimmune illnesses [18]. In a patient with resistant MuSK-positive MG, bortezomib was tested with considerable improvement; however, a significant confounding factor was that the patient had received RTX 19 days before starting bortezomib. Although further research is needed, boratezomib may show promise in MG [8].
While there is class I evidence supporting the short-term use of IVIG in acute worsening or myasthenic crisis, data supporting maintenance therapy is less strong and limited to class III evidence (107–109). Immunoglobulins (Ig) have broad-spectrum immunomodulatory actions and influence the body through a variety of B-cell, T-cell, complement, and Fc receptor-modifying mechanisms. IVIG is a helpful treatment option when a quick response is needed in worsening or poorly controlled MG [8]. SCIg (2 g/kg) was given to 23 persons with deteriorating mild or moderate MG over the course of 4 weeks in a prospective open-label trial. When compared to baseline at weeks 2, 4, and 6, the main result was a substantial decrease in QMG scores at 6 weeks. The most frequent adverse effects, which affected 74% and 61% of individuals, respectively, were headache and infusion responses. This research's QMG score improvement was similar to that of the IVIg pivotal study [9].
DISCUSSION:
The treatment of myasthenia gravis has been greatly enhanced by recent developments in immunotherapy, especially for individuals who do not respond to traditional therapies. By stopping complement-mediated damage at the neuromuscular junction, complement inhibitors like eculizumab and zilucoplan have shown remarkable effectiveness. Long-term therapeutic requirements and patient response variability, however, continue to be significant issues.
Efgartigimod and other FcRn antagonists are a potential strategy since they lower the levels of pathogenic IgG in the blood. Although long-term safety data are currently being assessed, clinical investigations have demonstrated a quick and consistent improvement in illness severity scores.
Overall, the expanding role of targeted immunotherapy in MG is supported by the available data. To maximize treatment approaches, future research should concentrate on large-scale clinical trials, long-term safety assessment, and cost-effectiveness.
CONCLUSION:
Most individuals with MG respond well to current immunotherapy, although there are serious side effects, delays in therapeutic effects, and worldwide immunosuppression as costs. Faster-acting, more precisely targeted immunotherapies that could enhance results and lessen side effects are promised by novel treatment strategies. While some innovative treatments will soon be used in clinical settings, others are still far off. Given the variety of MG treatment options available, cost becomes a significant consideration, and in many situations, less expensive medications may be deemed desirable. To determine the cost-effectiveness of innovative treatments in comparison to more conventional options, health economic studies are required.
ACKNOWLEDGEMENT:
We express our sincere gratitude to the management of JKK Munirajah Institute of Health Sciences College of Pharmacy for providing the necessary facilities and support to carry out this study. We would like to extend our heartfelt thanks to our guide, Ms. K.P. Anagha, Assistant Professor, Department of Pharmacology, for her continuous guidance, encouragement, and valuable suggestions throughout the preparation of this study.
REFERENCES

Nikhil Khopade*, Kakasaheb kore, Sucheta bhise, Adinath bhusari, Dr. Rajkumar Shete, Curcumin Phytosomes: A Promising Nanocarrier System for Enhancing Bioavailability and Therapeutic Efficacy, Int. J. of Pharm. Sci., 2025, Vol 3, Issue 5, 2448-2460. https://doi.org/10.5281/zenodo.15425503
 10.5281/zenodo.14950319
10.5281/zenodo.14950319