
We use cookies to ensure our website works properly and to personalise your experience. Cookies policy
1,2,3,4,5 Bharati Vidyapeeth College of Pharmacy, Kolhapur, Maharashtra, India.
6 Govindrao Nikam College of Pharmacy, Sawarde, Maharashtra, India.
Data integrity is a core component of pharmaceutical quality systems, underpinning regulatory compliance, product reliability, and patient safety. With the increasing digitalization of manufacturing and quality control processes under the Pharma 4.0 paradigm, governance of electronic data has become central to lifecycle-based quality assurance. This review critically examines recent developments (2024–2025) in global data integrity expectations, focusing on guidance and enforcement trends from the United States Food and Drug Administration (FDA), the European Medicines Agency (EMA), the Medicines and Healthcare products Regulatory Agency (MHRA), and the World Health Organization (WHO). A structured evaluation of regulatory guidelines, draft revisions including EU GMP Annex 11 and Annex 22, inspection observations, and recent scientific literature was undertaken to identify emerging compliance themes and systemic vulnerabilities in pharmaceutical manufacturing and quality control settings. The expanded ALCOA+ principles and evolving governance-oriented interpretations are discussed in relation to audit trail management, computerized system validation, and quality culture. Common deficiencies reported in warning letters such as incomplete raw data retention, inadequate audit trail review, and weak access controls are analyzed from a risk management perspective. Emerging technologies, including blockchain-based traceability and deterministic artificial intelligence applications, are considered with respect to regulatory feasibility and implementation challenges. This review provides a consolidated framework to support sustainable, inspection-ready data governance in modern pharmaceutical environments.
In pharmaceutical manufacturing, documented evidence has always formed the basis of regulatory compliance and product quality assurance. However, the traditional maxim “not documented, not done” has progressively evolved into a stricter expectation: data must be complete, reliable, and attributable throughout its lifecycle to be considered credible evidence of compliance.1 Data integrity therefore extends beyond record-keeping; it represents the assurance that manufacturing, testing, and release decisions are supported by accurate and trustworthy information. Within modern Pharmaceutical Quality Systems (PQS), data serve as both operational inputs and regulatory evidence, directly influencing patient safety and market authorization decisions.1
Historically, regulatory attention to data reliability emerged through the Application Integrity Policy (AIP) and the implementation of 21 CFR Part 11, which established requirements for electronic records and electronic signatures.3 While these frameworks initially focused on technical controls within computerized systems, contemporary regulatory expectations have shifted toward a lifecycle-based governance model. This approach integrates data generation, processing, review, archival, and retrieval within a structured quality risk management framework. Regulatory authorities increasingly regard data as a surrogate indicator of product quality; deficiencies in data integrity are therefore interpreted as potential risks to product safety, even when physical test results appear acceptable.5
Recent inspection trends underscore the persistence of systemic data integrity deficiencies across global manufacturing networks. Enforcement activities during 2024–2025 demonstrate a continued emphasis on audit trail review failures, inadequate raw data retention, shared system credentials, and weaknesses in computerized system validation.7 Rather than focusing solely on isolated documentation errors, regulators are increasingly identifying broader governance failures within quality systems. This evolution reflects a transition from reactive compliance monitoring to risk-based, intelligence-driven inspection strategies.7
The globalized nature of pharmaceutical supply chains further complicates oversight. Multinational manufacturing arrangements, reliance on contract laboratories, and cross-border data hosting environments introduce variability in technological maturity and quality culture.9 Regulatory agencies have consequently adopted enhanced inspection tools, including remote record requests under Section 704(a)(4), expanded reliance on data analytics, and targeted surveillance mechanisms to identify high-risk facilities.7 These developments signal a broader regulatory shift toward digital scrutiny and proactive risk identification.
Despite the extensive availability of regulatory guidance, there remains a need for an integrated analysis that synthesizes recent enforcement trends with evolving digital compliance frameworks, particularly considering the 2024–2025 draft revisions to EU GMP Annex 11 and the introduction of Annex 22 addressing artificial intelligence. Current literature often examines data integrity principles, warning letters, or digital transformation independently, but fewer analyses critically evaluate how these elements intersect within contemporary manufacturing and quality control environments.
Accordingly, this review aims to examine the evolving global regulatory landscape governing data integrity in pharmaceutical manufacturing and quality control systems. By synthesizing regulatory guidelines, draft revisions, warning letter observations, and recent peer-reviewed literature, the article provides a structured analysis of integrity principles, systemic root causes of violations, and practical implementation challenges within hybrid and digitally integrated environments.7
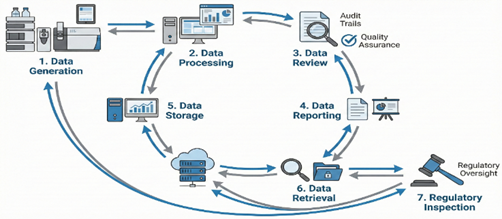
Fig 1: Lifecycle model of data integrity governance in pharmaceutical manufacturing and quality systems.
2. CONCEPT OF DATA INTEGRITY AND ALCOA+ PRINCIPLES
Data integrity in pharmaceutical systems refers to the assurance that data are complete, consistent, accurate, and reliable throughout their entire lifecycle from initial generation to long-term archival and retrieval.5 Within regulated environments, integrity extends beyond data correctness; it encompasses the governance structures, technical controls, and organizational behaviors that ensure data remain trustworthy under inspection and throughout product lifecycle management. As pharmaceutical operations increasingly rely on electronic and hybrid systems, integrity expectations have evolved from basic documentation practices to structured lifecycle control models.
To operationalize these expectations, regulatory authorities and industry bodies adopted the ALCOA framework as a practical interpretative tool for evaluating GxP data reliability.4 Over time, the framework has expanded to ALCOA+ and, more recently, to the conceptually broader ALCOA++ model, reflecting the increasing complexity of digital infrastructures and organizational accountability.
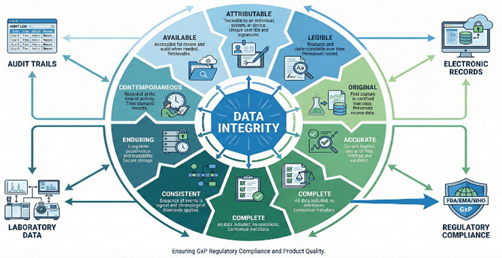
Fig 2: ALCOA+ principles framework for ensuring pharmaceutical data integrity.
2.1 The Foundational ALCOA Framework
The original ALCOA principles Attributable, Legible, Contemporaneous, Original, and Accurate remain the primary evaluative lens through which inspectors assess compliance with data integrity expectations.
Attributable requires that each data entry be traceable to a specific individual, along with the time and context of its generation.10 In computerized systems, this principle necessitates unique user credentials, controlled access rights, and audit trail functionality capable of recording user activities in a tamper-evident manner.4 The continued presence of shared logins in inspection findings underscores the centrality of this requirement.
Legible ensures that records remain readable and understandable throughout their required retention period, which may extend over several decades, particularly for stability or biologics data.10 In electronic environments, legibility also implies format sustainability and compatibility across evolving software platforms.
Contemporaneous documentation requires that data be recorded at the time the activity is performed.4 Delayed transcription, memory-based recording, or retrospective completion of records compromises temporal accuracy and remains a frequent observation during inspections.
Original data refer to the first capture of information or a verified true copy that preserves associated metadata, including timestamps and audit trail history.4 In hybrid systems, preserving originality can be challenging when electronic outputs are manually transcribed into paper records, increasing the risk of discrepancies.
Accurate data must reflect the true observation or measurement without manipulation, selective omission, or unjustified alteration.10 Where corrections are necessary, they must be traceable, justified, and transparent, preserving both the initial and modified entries.
Collectively, the ALCOA principles provide a structured foundation; however, they primarily address data characteristics rather than broader governance behaviours.
2.2 Expansion to ALCOA+
As digitalization advanced, regulators recognized that the original five attributes did not fully capture lifecycle control requirements. The expanded ALCOA+ framework incorporates four additional attributes: Complete, Consistent, Enduring, and Available.10
The ALCOA+ expansion reflects a shift from isolated data-point evaluation toward full lifecycle integrity management.
2.3 Emerging ALCOA++: Governance and Quality Culture
The emerging concept of ALCOA++ extends beyond technical attributes to include elements such as traceability and transparency within organizational systems.4 Although not formally codified in regulatory text, this conceptual evolution recognizes that technical controls alone are insufficient to prevent integrity failures.
ALCOA++ situates data integrity within a broader governance ecosystem, where management oversight, quality culture, and accountability mechanisms determine whether principles are consistently applied. In this view, data integrity is not merely a system property, but a behavioral outcome shaped by leadership priorities, workload pressures, training adequacy, and ethical climate.
This broader interpretation aligns with contemporary regulatory emphasis on senior management responsibility and quality culture as foundational elements of sustainable compliance. Rather than treating integrity as an isolated IT concern, ALCOA++ frames it as an integrated quality system function.
2.4 Practical Translation of ALCOA+ Principles into System Controls
The table below summarizes current expectations and typical implementation mechanisms aligned with contemporary regulatory interpretations.
|
Principle |
Core Expectation in 2025 |
Practical Implementation Mechanism |
|
Attributable |
Explicit linkage of each action to a unique user identity |
Unique credentials; prohibition of shared accounts; biometric authentication where appropriate.4 |
|
Legible |
Sustainable readability across technological transitions |
Use of standardized, non-proprietary formats (e.g., PDF/A); validated archival systems.4 |
|
Contemporaneous |
Real-time, system-controlled timestamping |
Network Time Protocol (NTP) synchronization; automated electronic capture.4 |
|
Original |
Preservation of first capture data and associated metadata |
Write-once-read-many (WORM) storages; secure audit trails.11 |
|
Accurate |
Faithful reflection of observed results |
Automated data capture (ADC); validated calculation tools.4 |
|
Complete |
Inclusion of all generated data, including repeats and failures |
System configurations preventing record deletion.1 |
|
Consistent |
Chronological and procedural coherence |
Integrated LIMS/ERP timestamps; structured audit trail review.4 |
|
Enduring |
Long-term data preservation |
Redundant storage; periodic restore testing.11 |
|
Available |
Prompt retrievability during inspection |
Centralized, searchable digital archives.4 |
3. REGULATORY EXPECTATIONS: THE 2025 GLOBAL FRAMEWORK
The regulatory landscape governing pharmaceutical data integrity has undergone notable evolution during 2024–2025, reflecting increasing digitalization of manufacturing systems and heightened scrutiny of computerized environments. Regulatory authorities have shifted from principle-based expectations toward more structured and operationally explicit guidance, particularly in relation to computerized system governance, cybersecurity, artificial intelligence, and risk-based inspection strategies. These developments collectively signal a transition toward digitally mature regulatory oversight.
3.1 United States Food and Drug Administration (FDA): Risk-Based Surveillance and Digital Oversight
The United States Food and Drug Administration (FDA) has continued to emphasize data integrity as a central component of Good Manufacturing Practice (GMP) compliance.7 Recent enforcement patterns demonstrate a sustained focus on systemic weaknesses rather than isolated documentation errors, particularly in areas involving audit trails, electronic record management, and access control mechanisms.7
A notable development in 2025 has been the reported implementation of internal analytical tools, including artificial intelligence–supported systems such as “Elsa,” designed to identify inspection priorities based on adverse event trends, Form 483 observations, and compliance data signals.7 While inspection authority remains grounded in statutory provisions, these analytical tools represent a move toward risk-based surveillance and data-driven targeting of facilities where patterns suggest potential governance deficiencies.
In parallel, the FDA has reiterated that compliance with 21 CFR Part 11 extends beyond software validation to encompass the broader ecosystem of people, processes, and controls interacting with computerized systems.3 Updated guidance communications reinforce the expectation that electronic records must be managed within a lifecycle framework incorporating access controls, audit trail review, backup verification, and validation of data processing functions. The 2025 Guidance Agenda further underscores emerging regulatory priorities including advanced therapeutic products and novel analytical methodologies which inherently depend on reliable digital data infrastructures.18
Collectively, these developments reflect a regulatory philosophy that integrates technological oversight with quality system accountability, reinforcing the principle that unreliable data compromise product quality assurance irrespective of final test outcomes.
3.2 European Medicines Agency (EMA): Revision of Annex 11 and Introduction of Annex 22
Within the European Union, recent draft revisions to EudraLex Volume 4 represent significant developments in the governance of computerized systems.14 The updated Annex 11 expands the scope and granularity of expectations related to lifecycle validation, cybersecurity, and access management, reflecting increasing regulatory attention to digital risk control.15
Among the notable revisions is the formalization of periodic system review requirements.14 Computerized systems are expected to undergo documented, scheduled evaluation to confirm continued suitability, validated state maintenance, and alignment with evolving regulatory expectations. This approach reinforces lifecycle validation rather than one-time qualification.
Cybersecurity considerations are also explicitly integrated into GMP expectations.14 References to frameworks such as NIS2 and ISO 27001 indicate that network protection, patch management, and vulnerability monitoring are no longer viewed solely as IT responsibilities but as components of quality system governance. In this context, cybersecurity failures may constitute GMP deficiencies if they jeopardize data integrity or system reliability.
Identity and access management controls receive particular emphasis. The draft guidance explicitly discourages shared accounts and highlights the necessity of robust authentication mechanisms that ensure traceable user activity.14 These measures directly address recurrent inspection findings involving unauthorized system access or inadequate attribution controls.
The introduction of Annex 22, focusing on artificial intelligence applications within GMP environments, represents an additional regulatory advancement.21 The framework differentiates between AI applications considered critical to product quality or patient safety and those categorized as non-critical. For critical applications, deterministic and explainable models are expected, with documented validation demonstrating performance equivalence to established processes.21 non-critical applications may incorporate more flexible AI tools, provided that human oversight mechanisms are clearly defined and documented.
Together, Annex 11 and Annex 22 signal a regulatory shift toward structured digital governance, where system validation, cybersecurity resilience, and algorithm transparency are integrated within GMP expectations.
3.3 MHRA and WHO: Emphasis on Data Governance and Quality Culture
The United Kingdom’s Medicines and Healthcare products Regulatory Agency (MHRA) continues to frame data integrity within a broader data governance model.23 Rather than focusing solely on technical controls, the MHRA emphasizes organizational accountability, procedural clarity, and management oversight as determinants of sustainable compliance.
A recurring theme within MHRA inspection communications is the rejection of “human error” as an insufficient root cause explanation.24 Investigations are expected to examine contributing systemic factors, including inadequate training, poorly designed interfaces, ambiguous standard operating procedures, or unrealistic operational timelines. This approach reinforces the principle that data integrity failures often arise from structural weaknesses rather than isolated individual lapses.
Similarly, the World Health Organization (WHO), through Technical Report Series (TRS 1033, Annex 4), highlights senior management responsibility in fostering an environment conducive to transparent reporting and ethical conduct.5 The WHO guidance stresses that employees must be able to report deviations or data concerns without fear of retaliation, as organizational culture directly influences the likelihood of intentional data falsification or concealment.2
Across these regulatory bodies, a consistent theme emerges sustainable data integrity requires integration of technical safeguards, procedural clarity, and ethical governance. The contemporary regulatory framework therefore extends beyond system validation to encompass leadership accountability and quality culture maturity as integral components of compliance. 2
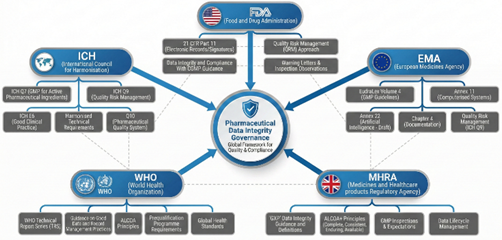
Fig 3: Global regulatory framework governing pharmaceutical data integrity.
4. COMMON DATA INTEGRITY VIOLATIONS IN MANUFACTURING AND QC LABORATORIES
A review of regulatory enforcement actions issued during 2024–2025 indicates that data integrity violations are seldom isolated procedural lapses; rather, they frequently reflect systemic deficiencies within quality oversight, computerized system governance, and organizational culture. Inspection findings across jurisdictions demonstrate recurring patterns, particularly within quality control laboratories, manufacturing documentation practices, and third-party testing arrangements.
4.1 Laboratory Control Violations
Quality Control (QC) laboratories continue to represent high-risk environments for data integrity breaches due to their reliance on computerized analytical instruments and complex data processing workflows. One frequently cited deficiency involves the “orphaning” of instrument data, where standalone workstations connected to chromatographic systems or spectroscopic instruments operate outside validated network controls.1 In such cases, insufficient access restrictions may permit deletion, modification, or selective reprocessing of analytical runs without adequate oversight.
Another common observation concerns failure to retain complete raw data.17 Regulatory authorities have identified instances in which only final processed results were preserved, while original chromatograms, spectra, or intermediate injections were deleted or overwritten. The absence of primary data prevents independent reconstruction of analytical events and compromises the ability to verify test authenticity. These practices directly contravene ALCOA+ expectations relating to completeness and originality.
Additionally, deficiencies in audit trail configuration or review processes remain prevalent. In several enforcement actions, audit trails were either disabled, inadequately configured, or not routinely reviewed prior to batch disposition decisions.1 Such omissions undermine traceability and weaken the evidentiary reliability of laboratory results.
4.2 Manufacturing and Production Documentation Failures
Within manufacturing operations, regulatory attention frequently centers on contemporaneous documentation and record authenticity.8 A recurring practice involves the temporary recording of process parameters such as weights, temperatures, or equipment settings on unofficial worksheets or scrap paper prior to transcription into formal batch records. This “uncontrolled document” practice introduces opportunities for transcription errors, delayed recording, and potential alteration of entries before final documentation.
Inspection findings have also documented retrospective completion of records, commonly described as backdating.12 When batch documentation is finalized after the fact rather than recorded at the time of performance, the reliability of manufacturing history becomes questionable. Missing signatures, incomplete logbooks, and inconsistent timestamps further compromise attribution and sequence reconstruction.
In some cases, spreadsheet-based calculations used in production or quality review processes were found to be unvalidated or insufficiently controlled.8 The use of uncontrolled electronic tools introduces risks of formula errors, hidden data manipulation, or inadvertent overwriting of values that may directly affect potency, yield, or specification assessments.
4.3 Third-Party Testing and Contract Laboratory Risks
An emerging trend in 2024–2025 enforcement activities involve increased scrutiny of third-party laboratories and contract testing facilities. Regulatory actions have identified instances of duplicated test results, falsified animal study data, and replication of analytical outcomes across unrelated studies.3 These cases illustrate that outsourcing analytical or nonclinical testing does not transfer regulatory responsibility from the sponsor or marketing authorization holder.
Sponsors remain accountable for ensuring that contract laboratories operate within validated systems and maintain robust data governance controls.3 Failure to adequately qualify and audit third-party facilities can result in enforcement actions against both the contractor and the contracting organization. The regulatory expectation is therefore not limited to contractual agreements but extends to ongoing oversight, data verification, and independent quality review.
The table below summarizes frequently cited violation categories and their regulatory implications during the 2024–2025 period.
|
Violation Category |
Specific Observation (2024–2025) |
Regulatory Implication |
|
Audit Trails |
Disabled, inadequately configured, or not reviewed during batch release. |
Inability to reconstruct events; compromised traceability.1 |
|
User Access |
Shared passwords or uncontrolled system privileges. |
Loss of attribution and accountability.4 |
|
Raw Data |
Deletion of trial injections or failed results. |
Violation of completeness and originality principles.1 |
|
Contemporaneousness |
Recording data retrospectively based on memory. |
Reduced reliability; increased risk of fabrication.12 |
|
Systems |
Use of unvalidated spreadsheets for calculations. |
Risk of undetected computational errors affecting product quality.8 |
|
Archival |
Poor storage practices; illegible or unstable records. |
Failure to meet enduring and legibility requirements.28 |
5. ROOT CAUSES OF DATA INTEGRITY FAILURES
Sustainable remediation of data integrity deficiencies requires analysis beyond observable deviations toward identification of underlying systemic contributors. Regulatory authorities consistently emphasize that isolated corrective actions such as retraining or disciplinary measures are insufficient when foundational governance weaknesses remain unaddressed. Root causes of integrity failures can broadly be categorized into technical, procedural, and organizational domains, although these categories frequently interact in complex ways.
5.1 Technical and Procedural Contributors
Technical vulnerabilities often originate from legacy computerized systems that were implemented prior to contemporary regulatory expectations for access control, audit trail configuration, and lifecycle validation.29 Older analytical instruments or standalone workstations may lack secure user authentication, granular permission controls, or immutable audit trail functionality. In such environments, reliance on procedural controls alone may be insufficient to mitigate manipulation risk.
Hybrid documentation workflows represent an additional vulnerability.14 When data are generated electronically but subsequently transcribed, reviewed, or approved on paper records, gaps may emerge between original capture and final documentation. This disconnect can create opportunities for selective transcription, delayed entry, or undocumented alterations, particularly if reconciliation processes are weak or inconsistently applied.
Procedural deficiencies also contribute significantly. Ambiguous standard operating procedures (SOPs), inadequate guidance on data review responsibilities, and insufficient clarity regarding audit trail expectations can lead to variable practices across shifts or departments.24 Inconsistent training programs and lack of role-specific accountability further exacerbate procedural drift, particularly in high-throughput manufacturing or laboratory settings.
Importantly, technical system design and procedural clarity are interdependent. A poorly configured system that permits unrestricted data deletion cannot be fully controlled through SOP language alone; conversely, robust software controls may fail if review responsibilities are not clearly defined and periodically verified.
5.2 Organizational and Cultural Determinants
Beyond technical and procedural factors, organizational culture frequently represents the most influential determinant of sustained data integrity performance. Regulatory communications increasingly highlight that pressure to meet production targets, release timelines, or commercial objectives may inadvertently incentivize documentation shortcuts or selective reporting.2 When employees perceive that productivity is prioritized over transparency, integrity risks increase.
The authority and independence of the Quality Unit (QU) play a central role in mitigating such risks.8 If quality oversight functions are structurally subordinate to operational management or lack sufficient decision-making authority, there may be reluctance to escalate deviations, challenge incomplete documentation, or delay batch disposition. Sustainable data governance therefore depends not only on formal structures but also on leadership commitment to ethical compliance.
Both the MHRA and FDA have cautioned against attributing recurring deficiencies solely to “human error.”24 In many cases, apparent individual mistakes are symptomatic of deeper systemic contributors, including excessive workload, inadequate staffing, poorly designed user interfaces, or insufficient system validation. Effective root cause analysis must therefore examine environmental and structural influences rather than focusing narrowly on individual behavior.
5.3 Structured Root Cause Analysis (RCA) in Data Integrity Investigations
Quality Risk Management principles, as outlined in ICH Q9(R1), emphasize systematic identification and evaluation of risk sources within pharmaceutical systems.36 When applied to data integrity investigations, structured Root Cause Analysis (RCA) tools facilitate differentiation between superficial symptoms and fundamental control failures.
Commonly applied methodologies include:
|
RCA Tool |
Strategic Application in Data Integrity |
Outcome for Quality Assurance |
|
5 Whys |
Iterative questioning to determine why an audit trail was not reviewed or why data were entered retrospectively. |
Identifies training gaps, unclear responsibility allocation, or unrealistic timelines.31 |
|
Fishbone (Ishikawa) Diagram |
Categorization of contributing factors into People, Process, Equipment, Environment, and Management. |
Provides a multidimensional understanding of systemic vulnerabilities.32 |
|
Failure Mode and Effects Analysis (FMEA) |
Prospective evaluation of potential data manipulation risks within computerized systems. |
Enables prioritization of controls based on severity, occurrence, and detectability.33 |
|
Fault Tree Analysis |
Logical mapping of event combinations leading to integrity breach. |
Identifies interacting technical and organizational contributors.33 |
The effectiveness of RCA depends on objectivity, cross-functional participation, and management support. Superficial analyses that default to retraining as the primary corrective action are unlikely to prevent recurrence. Instead, sustainable remediation often requires engineering controls (e.g., system reconfiguration), workflow redesign, or organizational restructuring to address systemic drivers.
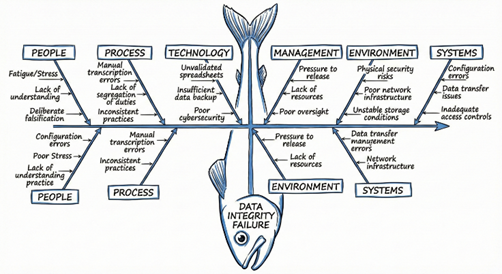
Fig 4: Root cause analysis of pharmaceutical data integrity failures using fishbone diagram.
6. DIGITAL SYSTEMS, ELECTRONIC RECORDS, AND AUDIT TRAILS
Contemporary data integrity assurance is intrinsically linked to the architecture, validation, and governance of computerized systems. As reflected in recent revisions to EU GMP Annex 11, computerized systems are no longer regarded merely as operational support tools but as regulated components of the Pharmaceutical Quality System (PQS).15 Their configuration, validation status, access management, and cybersecurity posture directly influence the reliability of GxP data.
6.1 Audit Trail Management and Review
An audit trail is defined as a secure, computer-generated, time-stamped record that enables reconstruction of events relating to the creation, modification, or deletion of electronic records.20 In modern regulatory practice, audit trails are not passive archival features but active compliance controls. For data impacting critical quality attributes or batch disposition decisions, authorities increasingly expect documented review of audit trail entries as part of routine quality oversight.29
Recent regulatory communications emphasize that audit trail review should be integrated into operational workflows rather than treated as an infrequent or purely technical task.20 This expectation reflects recognition that delayed review reduces the ability to detect unauthorized modifications, repeated test injections, or unexplained parameter changes in a timely manner.
However, large-scale digital operations generate substantial data volumes.34 In high-throughput analytical laboratories or automated manufacturing systems, the quantity of audit trail entries may render comprehensive manual review impractical. As a result, firms are increasingly implementing risk-based audit trail review strategies.29 Such approaches prioritize review of critical parameters and exception-based triggers (e.g., result changes, deletion attempts, privilege escalations), while lower-risk routine system events may be monitored through automated controls or periodic sampling.
The effectiveness of risk-based audit trail review depends on clear documentation of risk rationale, predefined review criteria, and evidence that exception thresholds are scientifically justified. Without these safeguards, selective review practices may inadvertently introduce oversight gaps.
6.2 Cybersecurity as an Integral Component of GMP
The integration of cybersecurity considerations into GMP frameworks represents a notable regulatory development.20 Increasing reliance on networked systems, cloud hosting environments, and remote access capabilities has expanded the potential attack surface for malicious activity. Ransomware incidents and unauthorized system intrusions have demonstrated that cybersecurity failures can directly compromise data availability, integrity, and traceability.
Recent regulatory revisions emphasize that cybersecurity controls must be embedded within validated system lifecycles.14 Expectations include documented infrastructure qualification, controlled network architecture, and ongoing monitoring of system vulnerabilities. Patch management programs must ensure timely deployment of security updates while maintaining validated system status.35
Furthermore, periodic penetration testing and vulnerability assessments are increasingly regarded as proactive risk mitigation measures rather than optional IT exercises.14 The integration of cybersecurity within GMP expectations underscores that data integrity encompasses protection against both internal manipulation and external compromise.
7. RISK-BASED APPROACHES AND CAPA IMPLEMENTATION
Effective management of data integrity risks requires systematic application of Quality Risk Management (QRM) principles, as outlined in ICH Q9(R1). The revised guideline emphasizes the importance of structured risk assessment methodologies that minimize subjectivity and ensure that control measures are commensurate with the potential impact on product quality and patient safety.36 Within the context of data integrity, this approach supports prioritization of resources toward systems and processes that present the highest risk of data compromise.
7.1 Designing a Risk-Based Data Governance System
A risk-based data governance framework requires classification of computerized systems according to both the criticality of the data generated and the vulnerability of those systems to manipulation, loss, or unauthorized modification.25 This classification facilitates proportional implementation of technical and procedural safeguards.
Systems categorized as high criticality and high vulnerability typically include analytical instruments such as High-Performance Liquid Chromatography (HPLC) systems used for batch release testing.1 Because the data generated directly influence product disposition decisions, these systems require comprehensive validation, controlled user access, routine audit trail review, and periodic verification of system integrity.
In contrast, lower criticality and lower vulnerability systems, such as certain environmental monitoring or facility management tools, may warrant less intensive oversight, including periodic review or sampling-based verification.1 However, even lower-risk systems must remain within the scope of quality system governance to ensure continued data reliability.
Risk categorization should be supported by documented justification and periodically reassessed to account for technological changes, evolving regulatory expectations, or process modifications. This lifecycle approach aligns with the principle that data integrity assurance must remain dynamic rather than static.
7.2 CAPA Effectiveness in Data Integrity Remediation
When data integrity deficiencies are identified, implementation of robust Corrective and Preventive Action (CAPA) is essential to prevent recurrence and restore confidence in affected systems. Regulatory inspections have frequently highlighted inadequacies in CAPA programs that rely primarily on retraining without addressing underlying technical or procedural contributors.12 Such superficial responses may correct immediate symptoms but fail to mitigate systemic risk.
Effective CAPA strategies typically incorporate multiple layers of intervention:
Engineering Controls:
Technical modifications to system configuration represent one of the most effective preventive measures. Examples include restricting data deletion privileges, enabling mandatory audit trail functionality, and implementing automated data capture to reduce manual transcription.24 These controls directly address system vulnerabilities that permit unauthorized data manipulation.
Administrative Controls:
Procedural improvements, including revision of standard operating procedures, clarification of documentation requirements, and enhanced personnel training, support consistent implementation of integrity controls.24 Clearly defined responsibilities for data review and approval reduce ambiguity and improve accountability.
Effectiveness Verification:
Demonstrating CAPA effectiveness requires objective evaluation using predefined quality metrics. Indicators such as reduction in late data entries, decreased frequency of audit trail exceptions, and improved compliance with documentation timelines provide measurable evidence of sustained improvement. Ongoing monitoring ensures that corrective measures remain effective over time. 12
8. PRACTICAL CHALLENGES IN IMPLEMENTATION
Despite the availability of comprehensive regulatory guidance, pharmaceutical organizations continue to encounter practical barriers in implementing fully compliant data integrity frameworks. These challenges often arise from technological limitations, infrastructure transitions, and operational constraints that complicate the integration of regulatory expectations into routine practice.
8.1 Legacy Systems and Hybrid Workflows
Many pharmaceutical facilities operate within technologically heterogeneous environments, where modern computerized systems coexist with legacy instruments lacking contemporary data integrity features.25 Older analytical platforms may not support audit trail functionality, secure user authentication, or automated data archival, requiring reliance on procedural controls such as manual verification or secondary review.
Hybrid workflows, involving electronic data generation followed by paper-based review or approval, present additional vulnerabilities.14 These workflows depend on accurate transcription, consistent reconciliation, and strict procedural discipline. Any breakdown in linkage between electronic source data and corresponding documentation may compromise traceability and increase the risk of transcription errors or undocumented modifications.
Replacement of legacy systems is often constrained by cost, operational disruption, and the need for extensive revalidation. Consequently, organizations must balance technical upgrades with interim procedural safeguards, increasing the complexity of maintaining consistent compliance.
8.2 Data Migration and Cloud-Based Infrastructure
The increasing adoption of cloud computing and Software-as-a-Service (SaaS) platforms introduces new governance considerations related to data control and regulatory responsibility. While external vendors may provide infrastructure and maintenance, regulatory accountability for data integrity remains with the regulated pharmaceutical organization.24
Data migration from legacy systems to cloud platforms presents particular risk, as incomplete transfer, loss of metadata, or improper validation may compromise historical data reliability. To mitigate these risks, organizations must implement structured migration protocols, validation testing, and verification of data completeness following transfer.
In addition, contractual arrangements with cloud providers must include clearly defined Service Level Agreements (SLAs), covering backup procedures, security controls, access management, and audit rights.15 Without adequate oversight, reliance on third-party infrastructure may introduce vulnerabilities that extend beyond direct organizational control.
9. CASE-BASED CRITICAL DISCUSSION
Evaluation of recent enforcement actions provides practical insight into how regulatory expectations are applied during inspections and highlights recurring weaknesses in data governance systems.
9.1 Case 1: Mid-Link Technology Testing Co., Ltd.
In 2024, regulatory investigators identified statistically improbable patterns in animal weight data submitted by Mid-Link Technology Testing Co., Ltd., where identical incremental changes were recorded across multiple subjects.39 Subsequent investigation revealed that technicians had entered estimated values when measurements could not be obtained reliably.39
This case illustrates the increasing use of data analytics by regulatory authorities to detect irregular patterns indicative of potential falsification. The regulatory response required comprehensive independent review of data integrity practices and removal of personnel involved in data manipulation.3 The findings underscore the importance of ensuring that recorded data reflect actual observations rather than inferred or estimated values.
9.2 Case 2: CCIC Huatongwei International Inspection Co., Ltd.
A 2025 warning letter issued to CCIC Huatongwei cited deficiencies in archival practices, including missing original records and inadequate documentation controls.28 Investigators also observed evidence of recent modification of electronic files and improper labeling practices.
This case highlights the critical importance of maintaining secure, permanent, and traceable records. Even when analytical work may have been performed correctly, failure to preserve original documentation undermines the credibility of the entire dataset and compromises regulatory confidence.
9.3 Case 3: Granules India Limited
Regulatory inspection of Granules India Limited identified deficiencies in deviation investigation and documentation related to equipment maintenance activities.30 The failure to adequately investigate and document potential process deviations demonstrated weaknesses in quality oversight.
This case illustrates that data integrity extends beyond laboratory results to include documentation of manufacturing operations and investigation processes. Incomplete or inadequate investigation records limit the ability to reconstruct events and assess potential impact on product quality.
10. FUTURE PERSPECTIVES: PHARMA 4.0 AND AUTOMATION
Advancements in digital technologies are reshaping pharmaceutical manufacturing, shifting data integrity management from reactive verification toward proactive and system-driven assurance. The concept of “digital maturity” emphasizes integration of automated data capture, real-time monitoring, and advanced analytics to minimize manual intervention and reduce opportunities for data manipulation or transcription errors. These developments, often associated with the Pharma 4.0 paradigm, offer potential benefits but also introduce new regulatory and validation considerations. 6
10.1 Blockchain and Distributed Ledger Technologies
Blockchain technology has emerged as a potential tool for strengthening data traceability and record security within pharmaceutical supply chains. By maintaining distributed, time-stamped records secured through cryptographic mechanisms, blockchain platforms can provide tamper-evident documentation of transactions across multiple stakeholders.6 This decentralized structure reduces the risk of unauthorized modification and enhances transparency in material tracking and batch history documentation.
Pilot initiatives, such as the MediLedger project, have demonstrated the feasibility of using blockchain to improve supply chain traceability and verification processes.6 However, integration of blockchain into regulated environments presents challenges, including system validation, scalability, and compatibility with existing regulatory frameworks. Additionally, ensuring controlled access, data privacy, and alignment with GMP documentation requirements remains essential for regulatory acceptance.
10.2 Pharma 4.0 and Real-Time Process Monitoring
The Pharma 4.0 framework promotes the use of interconnected sensors, automation platforms, and data analytics to enable real-time process monitoring and control.16 Internet of Things (IoT) technologies allow continuous acquisition of process parameters, including temperature, pressure, and environmental conditions, with direct integration into validated data management systems such as Laboratory Information Management Systems (LIMS) or Manufacturing Execution Systems (MES).
Automated data capture reduces reliance on manual recording and can improve data consistency and completeness.16 Furthermore, advanced analytical tools, including machine learning algorithms, may assist in detecting process deviations, identifying trends, and supporting predictive maintenance activities. These capabilities enhance process understanding and support proactive quality management.
Nevertheless, implementation of automated monitoring systems requires comprehensive validation to ensure data accuracy, system reliability, and regulatory compliance. Improper configuration, inadequate validation, or insufficient oversight may introduce new risks rather than eliminating existing ones.
10.3 Artificial Intelligence and Regulatory Considerations
Artificial intelligence (AI) applications are increasingly explored for functions such as visual inspection, process optimization, and data analysis. Regulatory discussions, including recent guidance developments, emphasize the importance of transparency, reproducibility, and explainability in AI-supported decision-making.21 Systems used in quality-critical applications must produce consistent and verifiable outputs and operate within defined validation parameters.
While AI tools may improve efficiency and support quality assurance activities, their implementation requires careful risk assessment, validation planning, and ongoing performance monitoring.21 Regulatory expectations emphasize that automated decision-making systems must remain subject to human oversight and quality review, particularly where product quality or patient safety may be affected.
CONCLUSION
Data integrity has evolved into a central component of pharmaceutical quality assurance, reflecting the increasing reliance on computerized systems and digital data throughout the product lifecycle. Regulatory developments during 2024–2025, including revisions to EU GMP Annex 11 and the introduction of Annex 22, demonstrate a growing emphasis on lifecycle governance of computerized systems, cybersecurity, and emerging technologies such as artificial intelligence. These changes reinforce the expectation that data reliability must be ensured through a combination of technical controls, procedural safeguards, and organizational oversight.
Analysis of recent inspection findings highlights that data integrity deficiencies often originate from systemic weaknesses, including legacy system limitations, inadequate procedural clarity, and insufficient quality oversight. Addressing these risks requires implementation of structured Quality Risk Management approaches, effective corrective and preventive actions, and integration of audit trail review, access control, and validation within routine quality system operations.
Emerging digital technologies under the Pharma 4.0 framework offer significant opportunities to enhance data traceability, reduce manual intervention, and support proactive quality monitoring. However, their successful implementation requires robust data governance frameworks supported by technical controls, structured quality risk management, and strong organizational commitment. As pharmaceutical manufacturing continues to evolve toward digitally integrated environments, maintaining complete, accurate, and reliable data remains essential for regulatory compliance, product quality assurance, and protection of patient safety.
ABBREVIATIONS
REFERENCES






Prathamesh Salunkhe, Chaitali Mane, Sanket Kumbhar, Venkat Battalwar, Koushal Kadam, Sanket Salunkhe, Data Integrity in Pharmaceutical Manufacturing: Evolving Global Regulatory Frameworks, Enforcement Trends, and Digital Governance Strategies, Int. J. of Pharm. Sci., 2026, Vol 4, Issue 2, 3799-3818. https://doi.org/10.5281/zenodo.18749504
 10.5281/zenodo.18749504
10.5281/zenodo.18749504