Fabtech College of Pharmacy, Sangola, Solapur, Maharashtra- 413307
Research-based practice are grounded in experimental investigations that provides safety as well as best practices for medical treatments. From original concept to implementation, the essential components of clinical trial design and development are examined in this review article. The different trial phases (I–IV), study design types (cohort studies, randomized controlled trials, adaptive designs), and methodological issues like blinding, randomization, sample size computation, and endpoint selection are all covered. Informed consent, Good Clinical Practice (GCP), and regulatory body oversight are among the ethical and regulatory frameworks that are included in the review. Along with new developments like decentralized trials and the use of digital technology, issues with recruitment, data management, and trial monitoring are covered. This study attempts to assist researchers, clinicians, and stakeholders in carrying out strong and morally sound clinical trials that significantly advance medical science and patient care by combining the most recent research and best practices.
The process of evaluating a medication or device's efficacy and safety in human beings is known as a clinical trial [1]. clinical trial is an organized procedure designed to determine if a medication or medical technology is safe and effective in treating, preventing, or diagnosing sickness either physiological abnormality [2].
Clinical trials are developed in stages, starting with early-phase studies that evaluate pharmacokinetics and safety (Phase I), moving on to larger trials that assess efficacy (Phase II), and concluding with confirmatory studies that compare interventions to standard care (Phase III). Phase IV, post-marketing surveillance, evaluates long-term efficacy and safety in practical contexts.[3] Clinical trials, and more especially randomised controlled trials [RCTs], continue to be the standard for comparing illness therapies, despite the fact that many study designs can achieve these objectives. Therefore, it is essential for healthcare professionals to comprehend the principles that underpin successful clinical trials in order to continue working in collaboration via care seeker or business to develop secure, highly potent treatments. It outline important ideas and the challenges involved in planning and carrying out a clinical research successfully.[4] Randomisation strategies, blinding procedures, study objectives, participant selection criteria, statistical power, and ethical considerations must all be carefully taken into account while designing a clinical trial. The integrity and safety of trial participants are further guaranteed by regulatory guidelines like the Indian Council of Medical Research guidelines, U.S.[5].

The moral basis of clinical research
Even though scientist 1753 work "A Treatise of the Scurvy" detailed earliest known contemporary interventional study, principled issues in person research are unresolved prior to mid twentieth era. Nuremberg Regulations established ten fundamental guidelines for human research in reaction the Nazis' illegal use of human beings in medical experiments during World War II[6]. The Nuremberg Code's informed consent principle, which had been advocated 30 years before, was ultimately explained in the Belmont Report. All study participants (with very few exceptions) must sign an informed consent form, which is now a requirement of clinical studies. It must explicitly state:[7]
In its most basic form, biomedical experiments aim for which observe individuals’ subjects' results under "experimental" circumstances that are managed by the researcher. This contrasts with noninterventional study designs, such as case-control and cohort studies, where the researcher measures the exposure of interest without influencing it. Because it enables randomisation of the intervention, a clinical trial design is frequently preferred. This substantially eliminates selection bias resulting from the imbalance of unknown or immeasurable confounders.
The ability to reveal causation in an RCT is one of these fundamental strengths[9].
Types of Clinical Trial
Advantages of clinical trials
Disadvantages of clinical trials
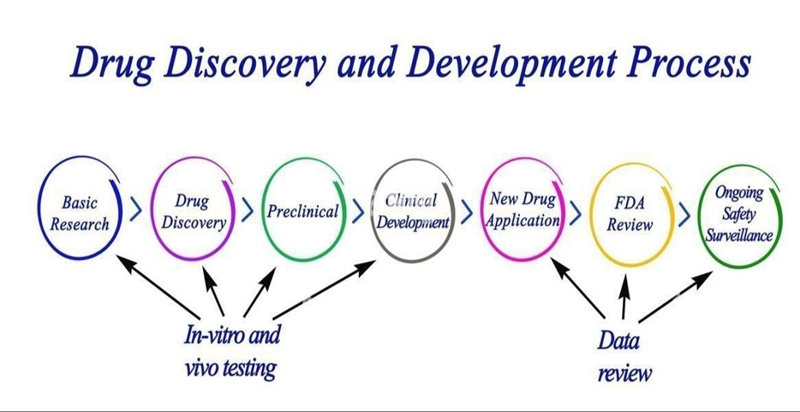
Phases of Diagnostics
Medical studies were carried out into stages, which is intended address a distinct research subject. Every clinical trial of a new medication or treatment goes through a number of stages to determine its efficacy and safety. The first step in a pharmacological investigation is the medication. design and the search for drugs test it on a small number of human volunteers before moving on to animal testing. If the trial is safe and beneficial, it can then be tested on a larger number of study participants. Cost and Time: The complete drug development process, from preclinical to phase IV, can take 12 to 18 years and frequently cost more than $1 billion[10].
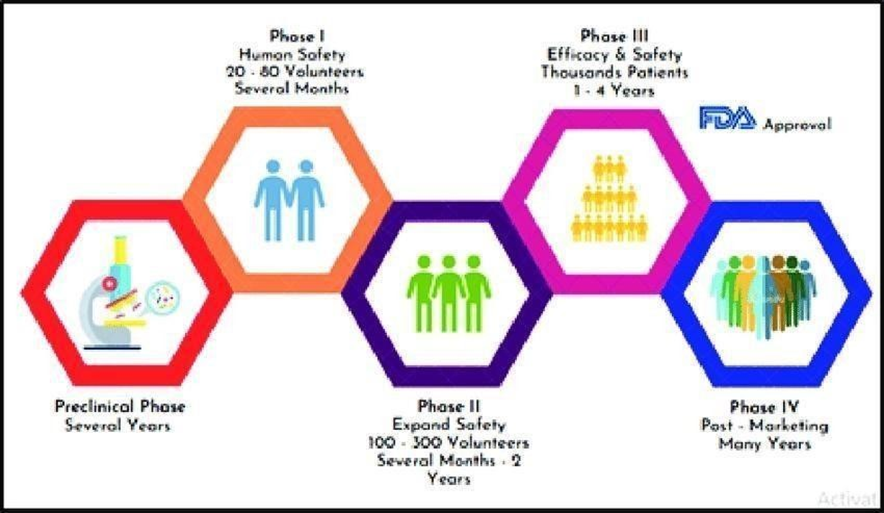
Pre-clinical study
Pharma industry gather primary efficacy, toxicity, and pharmacokinetic data through preclinical research, which includes within living organism as well as in the laboratory using broad range for dosage, before starting clinical trials of a medication. These tests are used by pharmaceutical companies to assess the scientific value of a medication. The decision of whether it requires additional development as a novel experimental medication is also necessary. [13,14]
Phase 0
Phase 0 is considered to be the trial's initial investigational launch. When the human trials were first being conducted, American FDA’s 2006.
Criteria for experimental IND analyses were are designed for producing a potential drug with the exact characteristics that preclinical research predicted. [13,15].
A single subtherapeutic medication is administered to a limited people. It may surprise you to learn that Phase 0 trials provide no specific information about the efficacy or safety of the test drug.
Additionally, pharmaceutical development companies conduct Phase 0 studies to choose potential drugs and ascertain the pharmacokinetic parameters in people for prospective development. [14]

Phase 1
A new drug's safety profile, higher allowable dosage, toxicity thresholds , PK also PD behaviour are the primary goals of phase I studies, which are the initial stage of human testing. Small cohorts of three to six patients are usually included in these studies, and doses are progressively increased until two or more patients experience intolerable toxicity at a certain dose level, indicating a DLT; the dose slightly below this threshold is known as the MTD. Because Phase I trials focus on safety rather than therapeutic benefit, participants typically include people with advanced, treatment resistant malignancies, regardless of tumour type. While pharmacodynamic evaluations aid in identifying the drug's biological effects on certain molecular targets, pharmacokinetic investigations offer details on the drug's absorption, distribution, metabolism, and elimination. PK–PD interactions are especially crucial for determining biologically efficacious dosages and ideal therapeutic exposure levels for molecularly targeted treatments. In general, safety, tolerability, MTD identification, and early signs of biological activity are the main outcomes of Phase I trials. [16,17,18]
Phase 2
To find out if a medication has anticancer activity at the ideal dose determined in Phase 1 studies, phase 2 trials conducted. Patients particular cancer types are enrolled in these studies; they are chosen Considering how the medication works, preclinical efficacy evidence, and any antitumor responses seen during Phase I. The tumour response rate, which indicates the extent of tumour shrinking in comparison to baseline data, is the main outcome of Phase II trials. In the past, tumour size was measured in two dimensions using the product of the longest diameter and its longest perpendicular diameter; a decrease in this value of at least 50% was regarded as an objective response. However, in order to guarantee consistent and repeatable evaluation, contemporary research employs the standardised Response Evaluation Criteria in Solid Tumours criteria, which depend on the longest diameter of target lesions. Treatment outcomes are classified by RECIST. especially for molecularly targeted therapies—are important outcomes in Phase II trials. .[19,20,21]
Phase 3
In vast and diverse patient populations, phase III clinical trials are intended to evaluate a novel treatment with the existing standard of care. In order to ensure that the results are broadly applicable, these trials are usually organised as randomised controlled trials (RCTs) with high sample sizes ranging from hundreds to thousands of participants. They are frequently carried out across numerous worldwide centres. Overall survival time, and the safety profile of the novel treatment in contrast to proven medicines are the Diseasefree interval (DFI), Healthrelated quality of life primary outcomes evaluated in Phase III trials. These phase 3 trials outcomes is crucial in deciding whether a medication should be included into clinical practice guidelines and approved by governing agencies like the food and drug administration or European agency. Fleming & Harrington's Counting Processes in Clinical Trials (1991), Pocock's Clinical Trials: A Practical Approach (1996), and the FDA's 2018 advice on oncology trial endpoints are all crucial resources for comprehending Phase III trial techniques.. [22,23,24]
Phase 4

After a medication has been approved by regulators and put on the market, stage 4 clinical studies, called after commercialization monitoring examination carried out. These trials are intended to track long-term safety, identify uncommon or delayed side effects, gauge practical efficacy, and analyse drug interactions or results in certain patient groups. These investigations could include more randomised trials, patient registries, or observational studies. Phase IV study results may lead to modified dose guidelines, new safety alerts, or, in extreme circumstances, the drug's removal from the market. The frequency of unusual side effects, long-term survival results, and actual treatment patterns are important objective. .[25,26,27]
Plans of clinical trials
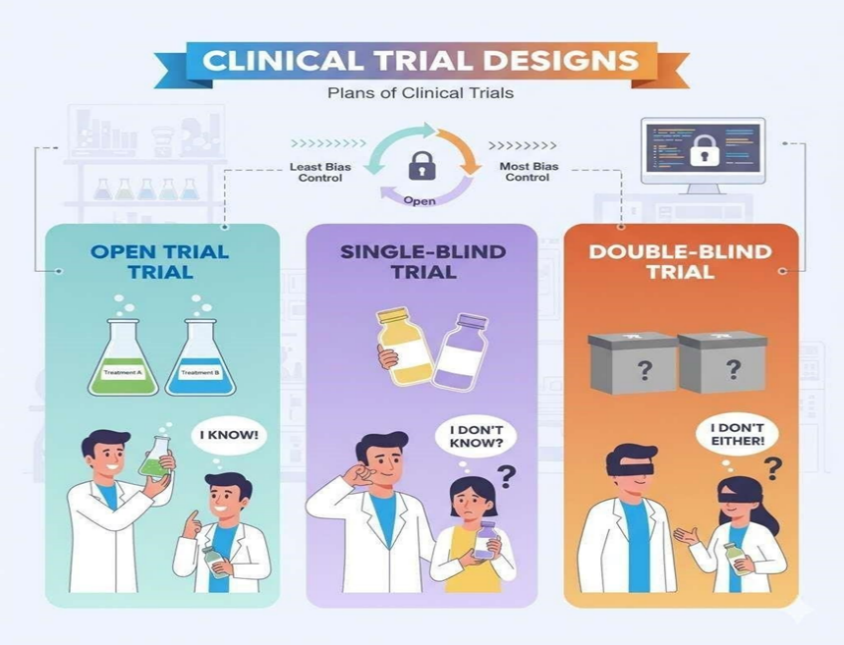
1. Open trial
Both the patient and the researcher are fully informed about the treatment in an open trial. The placebo effect is not lessened by these experiments, and they are subject to bias challenges. But occasionally they can't be avoided because it's not always possible to administer placebo treatments (see Blinding). Typically, bioequivalence studies employ this type of study design.
1. Sightless trial
A. Single sightless trial
In a single-sightless experiment, patient is Unknowing of specifics of the treatment, but the researcher is. There may not be a placebo effect as the patient is unaware of which treatment— the new one or another—is being given. In actuality, the researcher may treat the patient differently or subtly give the patient vital treatment-related information since he is aware of it, which could affect the study's findings.
B. Double blind trial
The 'new treatment' or 'old treatment' is assigned a set of numbers by a single researcher in a double-blind trial. The numbers are provided to the second researcher, but they do not specify to whom they have been assigned. Additionally, the age and sex distribution of patients is frequently more accurate in this approach. Because they typically produce the most reliable results, double blind (or randomized) experiments are therefore recommended.
C. Triple blind trial
Although the definition of triple blind may differ depending on the specific study design, some randomized controlled trials are regarded as such. Most commonly, this means that the person giving the treatment (usually a pharmacist), the researcher, and the participant are blind to what is being provided. Another possibility is that the statistician, researcher, and patient are blinded. It is possible that the response monitoring team is not aware of the intervention being administered to the study and control groups. The more widely used term "double blind trials" typically incorporates these extra measures; hence the term "triple blinded" is rarely used. To prevent anyone directly connected with the study from unduly influencing the study results, it implies an extra layer of protection.[28]
Design aspect clinical trials
Randomization Method
A) Randomized controlled study
Medical professionals can use high-level data from randomized control studies because of their extremely reliable design. The name of the control group suggests that it is a comparison group for the interventional group. The interventional group in this study receives the interventional drug, while standard set is given non active agent either the previous therapeutics. Trials might be blind, double-blind, or open.[29]
Newer Drug Designs
Adaptive randomization method
This paradigm works best when the expected impact size being assessed is big and is only applicable to studies with binary outcomes. The goal of the dropthe-loser and playthewinner designs is to increase the likelihood that patients will be randomized to the group that has the best chance of success. Based on the outcomes with prior patients, the likelihood of being randomly assigned to one or another group is adjusted. Following therapy, each patient's response is crucial in determining the research population's following makeup. The "play the winner" design randomly assigns more research participants for successful treatment. The "drop the loser" technique, research participants , taken out of the intervention arm that didn't work. The benefit is more opportunities for recruitment and greater exposure of individuals to an effective intervention; however, this may also lead to uneven group sizes, which may compromise statistical power. In certain trial scenarios, the following techniques can be coupled to form the basis of the adaptation.
a) Response Adaptive
This design requires rapid, quantifiable results but lowers enrollment to unsuccessful arms. It is inappropriate for illnesses with protracted recovery periods. As the trial goes on, it might introduce selection bias and impair allocation concealment. Temporal drift, or changes in patient or therapy characteristics over time, might potentially have an impact.
b) Ranking and selection
In the first stage of this adaptive design, participants were randomized to a variety of therapies as well as a placebo. Phase II uses a randomized parallel or adaptive design to evaluate best treatment from Phase 1 with a placebo. The final comparison compares all subjects who received the chosen intervention to all individuals who received a placebo during both periods. In situations with small sample sizes, it works well for comparing various interventions. Nonetheless, there is a possibility that the trial results will be tainted by the incorrect choice of the most effective treatment during phase I.
c) Sequential adaptive design
When the end point of efficacy, safety, or futility is reached, this design permits recurrent interim analysis and stopping. Unlike typical trials, a sequential trial does not know from the beginning how many volunteers will be required. When the first intermediate analysis satisfies predetermined ending criteria, the trial is terminated, potentially reducing the number of individuals exposed to a treatment that is previously proven effective or that is subpar, hazardous, or inutile. After a set or variable number of patients (group sequential) or after every patient (continuous sequential), analysis can be carried out. This design works best when treatment results happen very soon after recruitment and study enrollment is anticipated to last for a long time. This allows outcomes to be assessed.
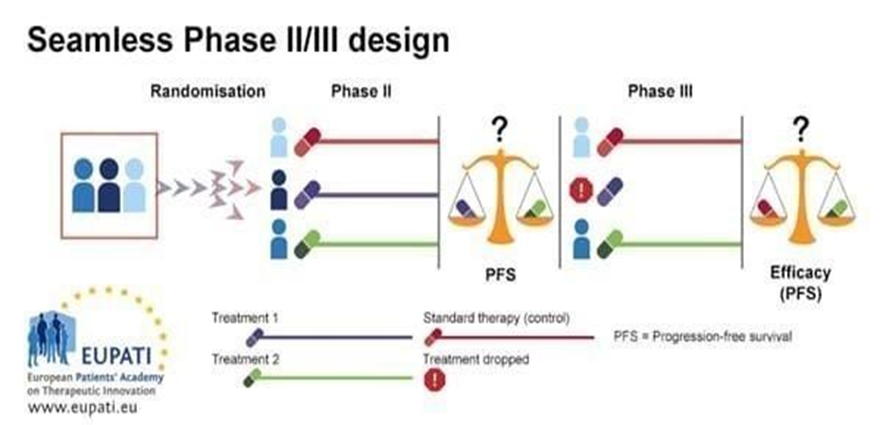
Seamless design
Smooth (Seamless Phase II/III) Design: A Brief Overview In a smooth or seamless design, the confirmatory phase of Phase III and the learning phase of Phase II are combined into a single continuous experiment. Initially, individuals randomly allotted for following trial groups:
Control A, B, or A+B. Ineffective arms are dropped by using an interim analysis. The Phase III confirmatory stage is carried out by the remaining effective arm or arms and control. Different kinds of seamless designs
Seamless inference: Phase II and Phase III data are processed jointly, and the same individuals continue.
Operational seamless: Although Phase II and Phase III are carried out using a single methodology, their data is examined independently.
Internal pilot design
Participants in a traditional Preliminary study are sometimes disqualified for analysis and cases in subsequent definitive investigations because of worries about training effects, selection bias, and carryover. When there are few patients, as in uncommon disease cases, assigning them to a Pilot trial instead of the final study may be viewed as an excessive strategy. An internal pilot study starts with a phase known as "pilot phase," and it continues until the study's sample size is reached (the definitive phase), while also analyzing the pilot subjects. Since internal pilots do not "use up" eligible patients or necessitate additional time or funding, they can be larger than external pilots.[31]
Ethical conduct
The proper regulatory agencies regularly monitor clinical trials. Before being authorized to conduct a study, any research including therapeutic as well as medical therapy in peoples have approval from directing ethical committee. How the local ethics council oversees noninterventional studies (observational studies or those that use data that has already been obtained) is up to them. This organization is known in the United States as the Institutional Review Board (IRB). A central (independent/for-profit) IRB may be used by investigators working at smaller institutions.
In order for research to be ethical, human participants must give their full and informed consent. Providing prospective patients with sufficient information about the clinical trial is one of the Rib's primary responsibilities. The patient's legally appointed representative may be asked for consent by researchers if the patient is incapable of giving consent on their own. In California, those who are capable of acting as the lawfully appointed representative have been given preference by the state.
Before conducting clinical trials, researchers and their staff in certain U.S. regions must obtain certification from the local IRB. HIPAA, the federal patient privacy law, and best professional practices must be understood by them. A collection of guidelines used globally for clinical trial conduct is called ICH Guidelines for Good Clinical Practice. Ensuring "rights, safety, and wellbeing of trial subjects are protected" is goal of the guidelines. The 1964 of WMA Declaration of Helsinki codifies guidelines for physicians to follow when doing clinical research.[32]
ICH GUIDELINES CLINICAL TRIALS
The basic principles of ICH:
ICH Guidelines
GCP are collection of international Responsible and Analytical standards for the appearance, operation, observation, auditioning, documentation, examination as well as documenting of clinical studies. Good clinical practices guarantee such trial subjects' legal powers, ethics, as well as privacy honored and secure, and that information and suspected results are accurate and reliable.[34]
Duty of chemist in therapeutic trials
Primary as well as main, we offer services essential for suitable storing of research base medical material, whether in a fridge either managed ambient temperature. This one way that pharmacists actively participate in research and clinical trials. Climate is monitored and recorded on a regular basis.

The pharmacist is also responsible for making confirm the IMPs constantly available also volunteer is given when prescribed. The proper use of the IMPs is explained to patients along with any written material that is given, including volunteer material pamphlet and aware Consigned document. To ascertain treatment compliance, patient IMP returns are tallied and recorded. Additionally, pharmacists will make sure administrated Investigational Medical Products is arranged according to trial's parameters as well as delivered accurately.
Oncology pharmacist sometimes oversees clinical trials and conducts research projects that try to improve the results of patients receiving chemotherapy or other supportive treatments like blood growth factor injections or antiemetics.
Chemist regularly execute analysis initiatives known Medication Utilization Judgment. In essence, providing information about how people utilize medications and tracking physician ordering routine. Since pharmacists confirm the drug applied correctly, Drug Utilization Evaluation frequently directed to medication inspection.
Pharmacists also conducted monitoring review to find out how doctors or patients feel about drugs. We use survey results to enhance the services we offer to our patients. Two surveys are now being carried out by NCC's oncology pharmacy.[36]
CONCLUSION
Clinical trials represent the final and most critical phase in function of developing new medicine, healthcare equipment, or therapeutic treatment. They provide the scientific foundation for determining modern therapies safe, therapeutic, and beneficial for human use. Conducted in multiple phases (Phase I–IV), each stage of a clinical trial serves a unique purpose from assessing basic safety and dosage to long-term effects and postmarketing surveillance.
REFERENCES



Sakshi Kengar, Dr. Savita Sonawane, Dr. Sanjay Bais, Design and Development of Clinical Trials, Int. J. of Pharm. Sci., 2025, Vol 3, Issue 12, 92-105. https://doi.org/10.5281/zenodo.17777727
 10.5281/zenodo.17777727
10.5281/zenodo.17777727
