SIMS College of Pharmacy
Lipid -based drug delivery systems are becoming increasingly popular as carriers of drugs due to their ability to overcome barriers to oral absorption. The purpose of this study was to prepare a novel formulation (lipid base) of a model drug Raloxifene HCL, a poorly water soluble drug using ‘DMPC’, ‘SoyPc’, and Labrasol. Lipid based drug delivery systems are prepared using conventional methods of preparation and without using any organic solvent during the process and finally converted into a solid intermediate for capsule filling. In addition, physical characterization studies using ‘DSC’, ‘XRD’, ‘SEM’. The formulation of Raloxifene HCL was prepared using (a) Drug, ‘DPMC’, ‘Soypc’ in the following combination 1:1:1. (a) 1:2 mixture of drug and phospholipids. Dissolution studies conducted in the buffers (Demineralized water, 0.1% polysorbate 80 in water, PH 1.2 / 3.0/ 4.5/ 6.8) using the usp type II dissolution apparatus showed an increased dissolution rate and better release profile . The optimized lipid based formulations was charged to accelerated stability studies for a period of two months .But there was a reduction in the release rate of Raloxifene HCLfrom the lipid based formulation showing intermediate stability which commands for the use of stabilizer in the formulation for attaining significant stability. Thus, the prepared lipid base formulation proved to be a potential technology for enhancing the transfer of poorly water soluble phospholipids compounds to the aqueous phase, thus enhancing bioavilibility.
Oral drug delivery remains the most popular route of administration. Indeed oral administration enhance patient’s quality of life because it is convenint and prevents intravenous drawbacks (catheter infection; thrombosis and extravasations)[19] .However, limitations in the physio-chemical properties of the drug sometimes prevent a successful therapeutic outcome. Specifically, problems of poor solubility and chemical stability in the gastrointestinal tract, poor permeability and sensitivity to metabolism are often causes that result in the rejection of potential drug candidates as commercial products [ 6] . Lipid-based delivery systems are gaining importance as carriers of drugs due to their ability to overcome barriers for oral absorption. The objective of the present study was to develop a Lipid Based Drug Delivery System for Raloxifene Hydrochloride to increase it saturation solubility and dissolution velocity for enhancing bioavailability while reducing variability in systemic exposure. RLX has low solubility in physiological pH conditions and extensive first pass metabolism are the major issues that has limited the therapeutic efficacy of Raloxifene HCL as an oral dosage form, which led to its low oral bioavailability of only 2 %. The approach used in this study is to formulate the poorly soluble drug, ( Raloxifene HCL as model drug) into encapsulated lipid dispersion which is converted into a solid intermediate by adsorbing with an inert hydrophilic carrier. The solid state characteristics of the granules shall be investigated with DSC, XRD and SEM studies. A capsule dosage form will be developed for incorporating the granulated lipid formulation containing the drug. The solubility and dissolution studies of the solid dosage form incorporating the granulated lipid formulation of Raloxifene HCL will be carried out in discriminating dissolution conditions and shall be compared with the commercially available Raloxifene hydrochloride tablet formulation (FIONA) and followed by assessment of stability.
MATERIALS AND METHOD
MATERIALS: Raloxifene hydro chloride, soy phosphatidyl choline , Dimyristoyl phosPhatidylcholine, Aerosil ® 200 pharma, Labrasol®, Tween-80, Propylene glycol , Anhydrous Glycol.
METHODS:
SOLUBILITY STUDIES:
The solubility studies for the drug were carried out using the Rotary shaking method at 200 rpm and addition of excess drug till saturation was observed for 48 Hrs. Then the samples were analyzed using HPLC at 287 nm. The solubility of the drug was calculated using the formulas:

Where,
Q = Percent of drug dissolved (% w/v); A =Standard Area; B = Test Area; C = Standard concentration (µg/mL) ;Wt. = Total weight of drug added; V1 = volume of test sample taken for dilution; V2 = diluted volume of test sample; V3 = volume of diluted sample (V2) taken for dilution; V4 = diluted volume of sample (V3) ; m = Amount/quantity (mg) of drug dissolved ; s = solubility ( mg/ ml) .
FORMULATION DEVELOPMENT
SELECTION OF EXCIPIENTS :
The excipients used in the formulation were selected based on their ability to enhance the solubility of drug. Similarly, the solubility studies were carried out for each of them.
Stage1: Screening of Phospholipids
Stage2: Selection of solvent, surfactant and co-surfactant
Stage3: Selection of Discriminating Dissolution medium
The selection of a discriminating medium was done based on the solubility of the drug in purified water and across pH buffers. Considering the drug’s highest and lowest solubility, the medium with optimum solubility of drug was selected. Based on the drug’s solubility in that medium, the conditions for discriminating dissolution of drug was fixed in included method, rpm, temperature, volume of medium, sink / non-sink conditions, time points for sample collection, etc. Table No.1 below gives various conditions for discriminating dissolution.
Table No 1: Dissolution condition for the Discriminating medium

Lipid formulation equivalent to unit dose (60 mg) of Raloxifene Hydrochloride was taken in a hard gelatin capsule (size 00) and the dissolution was carried out according to the above conditions. 10 mL aliquots of samples were withdrawn at pre-determined time intervals which were filtered through 0.45 µ PVDF (13 mm) filter. Then 1 mL of this filtered sample was diluted to 10 mL using diluent and were analyzed using HPLC. The cumulative percent drug dissolved at various intervals was calculated and plotted against time.
PROTOTYPE FORMULA DEVELOPMENT:
The selection and optimization was done based on the dissolution enhancement ability of the excipients used at different stages of formulation development. Finally the liquid formulation was converted into a solid intermediate by adsorbing with a solid carrier. It needs to be converted into an oral dosage form with high patient compliance. So, hard gelatin capsule of size ‘00’ was selected to deliver the drug product weighed equivalent to unit weight of the formulation (equivalent to 60 mg of Raloxifene HCl) and its in vitro characteristics were evaluated.
OPTIMIZATION OF FORMULA:
The formula was optimized by altering the concentrations of Labrasol, propylene glycol, Tween-80 and Aerosil 200 to obtain a formulation with enhanced dissolution profiles when compared to that of API and FIONA. The unit fill weight of the formulation was also taken into consideration for optimizing the formula. See Table No.2, indicates the variations in the concentrations of excipients for optimizing the formula. A brief explanation for each of the formula is given below.
Table No.2: Optimizing concentrations of solvent and surfactant

FORMULATION:
RLX-1:
In this formulation only Labrasol was used along with the drug and phospholipids. The resulting liquid formulation was taken in a capsule (equivalent to 60 mg of drug) and its dissolution studies were carried on.
RLX-2:
The effect of Lactose as an adsorbent was studied in this formulation.
RLX-3:
The effect of Aerosil 200 as an adsorbent was studied in this formulation.
RLX-4:
In this formulation, Propylene glycol was used as a solvent. Based on the previous studies Aerosil 200 was selected as the adsorbent and continued further.
RLX-5:
In this fomulation, the effect of Tween 80 as a co-surfactant was studied.
RLX-6:
In this formulation, the combined effect of Propylene glycol and Tween 80 was studied.
RLX-7:
In this formulation, the quantity of Labrasol, Propylene glycol and Tween 80 was altered in order to reduce the fill weight of the formulation.
RLX-8:
In this Aerosil 200 was not used and the dissolution studies were carried out for the optimized liquid formulation.

Fig. No.1: Flowchart for manufacturing of solid lipid based Formulation
EVALUATION OF FORMULATION :
EVALUATION OF BULK GRANULES:
This includes the evaluation of the flow properties(studied by angle of repose,car index,hausner’s ratio) and the solid state characterization of the bulk granules( studied by SEM,DSC, X – Ray Diffractometry (XRD).
Differential Scanning Calorimetry (DSC)
This gives us the information about the thermal properties of the sample like crystalline or amorphous nature, entrapment of drug with lipids and transition temperature of Phospholipids. The thermal properties were investigated using DSC Q – 1000, Differential scanning calorimeter with a refrigerated cooling system.
Process:
Weighed samples of 3 – 5 mg were taken in an open aluminum DSC pans and then sealed and crimped. These samples were scanned at a ramp of 10º C / min over a range of 10 – 300 º C. The samples included API, Placebo and Formulation.
X – Ray Diffractometry (XRD)
Process:
The x – ray diffractograms were recorded with approximately 500 mg of sample, using Xpert Pro X – Ray diffractometer with Cu K alpha as the source of radiation. Standard runs using a 40 KV voltage, a 40 mA current and a scanning rate of 0.02º min-1 over a 2–Theta range of 3–45º was used. The samples included API, Placebo and Formulation.
Scanning Electron Microscopy (SEM)
Process:
Few mg of sample was taken on the aluminum stub, present on the specimen holder. An adhesive was used to attach the sample to the stub. Now, this stub along with the sample was placed in the Hitachi vacuum evaporator and sputter coated with gold. This gold coated sample was placed in the chamber of SEM instrument for analysis. An accelerated voltage of 10 KV and a vacuum of less than 1 Pascal was used during the SEM analysis. Photographs were taken for each of the sample.
EVALUTION OF CAPSULE DOSAGE FORM
The formulated capsules were subjected to the following quality control tests.
Weight Variation Test:
Disintegration Test:
The in-vitro disintegration time was determined by using Disintegrating apparatus. A capsule was placed in to each of the six tubes of the apparatus and one disk was added to each tube. De mineralized water was used as the medium and maintained at 37º C. The time was recorded after completion of the disintegration of the entire capsule.
Percent Drug content:
This test was done by taking 4 capsules and the individual weights were calculated. Those capsules were dropped in a 1000 mL volumetric flask.
Where,
At = peak area of Raloxifene hydrochloride in the sample preparation, As = peak area of Raloxifene hydrochloride in the standard preparation, Sw = weight of Raloxifene hydrochloride working standard taken in mg, Tw = weight of sample taken in mg, P = Potency (%) of Raloxifene hydrochloride working standard calculated as API, Avg. wt = Average weight of the capsule taken in mg, L = label Claim of Raloxifene hydrochloride working standard calculated as API.
Dissolution:
The dissolution was carried out in discriminating conditions as mentioned above. In this the capsule representing unit dose of the drug was taken and dropped in the dissolution bowl, which contained 500 mL of de mineralized water and maintained at 37 ± 0.5º C. At pre determined time intervals of 15,30,45 min, 1, 2 and 3 Hrs. Aliquots of sample(10 mL) was withdrawn and was filtered through 0.45µ PVDF (13 mm) syringe filter.
This diluted sample was analyzed at 287 nm using HPLC. Then, cumulative % drug dissolved was calculated at each interval. A graph containing cumulative % drug dissolved on Y-axis and Time on X-axis was plotted.
Where,
At = peak area of Raloxifene hydrochloride in the sample preparation, As = peak area of Raloxifene hydrochloride in the standard preparation, Sw = weight of Raloxifene hydrochlorideworking standard taken in mg, MV = volume of the dissolution medium used in mL, L = label Claim of Raloxifene hydrochloride working standard calculated as API, V1 = Volume of sample taken for dilution (mL), V2 = Volume of diluted sample (mL), P = Potency (%) of Raloxifene hydrochloride working standard calculated as API.
STABILITY STUDIES :
Accordingly, the formulations were packed in HDPE bottles and charged for stability studies at 25º C / 60 % RH and 40º C / 75 % RH for a period of 2 months. At predetermined intervals of 15, 30 45 and 60 days, the samples were analyzed for the percent drug content and the drug dissolution rates. The dissolution studies were carried out in the developed 100 % release medium. These diluted samples were analyzed at 287 nm using HPLC. Then, cumulative % drug dissolved was calculated at each interval. A graph containing cumulative % drug dissolved on Y-axis and Time on X-axis was plotted and a comparison was done between the formulation and the marketed product (FIONA) regarding the stability criteria.
RESULTS AND DISCUSSIONS
SOLUBILITY STUDIES :
The solubility studies were carried out by adding excess drug to specific volume of water and agitated for 48 hrs at 200 rpm in a Rota shaker. The saturated samples were filtered and analyzed using HPLC.
SOLUBILITY IN PURIFIED WATER :
The solubility of Raloxifene HCl was investigated in purified water at both 25º C and 37ºC using the Rotary shaking method. The volumes of water and amounts of drug used for the solubility studies and its results are shown below in Table No. 3 and 4, respectively.
Table No 3: solubility studies of Raloxifene hydrochloride in water

Table No 4: solubility data of Raloxifene hydrochloride in water

The results revealed the low solubility of the drug in water. When compared to the solubility at 25º C, the solubility was more at 37º C indicating that temperature had an effect on the solubility of drug. Hence, it can be stated that Raloxifene HCL is a poorly water soluble drug.
SOLUBILITY ACROSS PH:
The solubility of Raloxifene HCl was also investigated in various buffers differing in their pH using the Rotary shaking method at 25º C. A pH – solubility curve for Raloxifene HCl was also plotted. The volumes of buffers and amounts of drug used are shown below in Table No. 5. The solubility results of drug in various buffers are shown below in Table No. 6.

Table No 5: solubility studies of Raloxifene hydrochloride across PH

Table No 6: solubility data of Raloxifene hydrochloride across PH

Fig No 2: PH Solubility profile of raloxifene Hydrochloride
Solubility data showed that at low pH, solubility was less and with increasing pH, solubility increased up to 3.0 and again solubility decreased at pH-8.0. So, it indicated that solubility of Raloxifene HCL is pH dependant. The dumb bell shaped curve in the pH solubility profile clearly indicated the drug’s highest solubility in 0.001 N HCL (pH-3.0) and least solubility in pH-8.0 buffer.
FORMULATION DEVELOPMENT
SCREENING OF PHOSPHOLIPIDS:
EFFECT OF PHOSPHOLIPIDS :
For this excess amount of drug was added to water in presence of certain quantity of phospholipids and was agitated on a Rota shaker for 48 hrs at 200 rpm at 25º C and 37 º C. The solubility data and results were shown below in Table No.7 and 8 respective.
Table No7: solubility studies for the effect of phospholipids on drug solubility

Table No 8: solubility data for the effect of phospholipids on drugs solubility

Based on the above results, Soy PC and DMPC showed better enhancement in drug’s solubility. Hence, for further studies in the formulation Soy PC and DMPC were selected.
EFFECT OF PHOSPHOLIPIDS CONCENTRATION ON SOLUBILITY OF THE DRUG:
In these studies instead of taking the phospholipids alone, both (selected phospholids) were taken in combination and their solubility enhancement was investigated. Certain ratios of drug: phospholipids were taken in 20 ml water and agitated for 48 hrs in a Rota shaker at 200 rpm. The solubility data and results were shown below in Table No. 9 and 10, respectively.
Table no 9 : solubility studies for the effect of phospholipid concentrations on drugs solubility
Table No 10: solubility data for the effect of phospholipids concentration on drugs solubility

The results showed an increase in the solubility with increasing concentrations of phospholipids. Hence, considering optimum quantities of phospholipids, the ratio comprising 1:1:1 of drug, Soy PC, DMPC respectively was selected and this ratio was extended further for formula development
SATURATION SOLUBILITY OF DRUG IN VARIOUS EXCIPIENTS:
For further enhancement of the drug’s solubility, few other excipients like solvent, surfactant and co-surfactant were investigated, which were selected initially based on the drug’s saturation solubility in the excipient. The solubility data and results of drug in various excipients were shown below in Table No. 11
Table No 11: solubility data and result of drug in various excipients

The above data showed good results for the drug’s solubility in that respective excipient. Of all, propylene glycol had the highest drug solubility and indicated its enhanced solubilisation nature for the lipophilic drug. Labrasol and Tween 80 also showed good solubility of drug indicating their surface activity property. The solubility of the drug in ethanol was due to its solubilisation property but it was not as good as compared to other excipients. Considering the above data, propylene glycol was selected as solvent, Labrasol as a lipid based surfactant and Tween 80 as a co-surfactant.
EFFECT OF EXCIPIENTS ON THE SOLUBILITY OF DRUG :
The solubility data and results for the effect of excipients on the solubility of drug are shown below in Table No.12. A graphical representation for the effect of excipients on the solubility of drug is shown below in Figure No.3.
Table No 12: solubility data for the effect of excipients on drugs solubility

Fig. No.3: Effect of excipient’s concentration on drug’s solubility

The above results showed enhanced solubility results with every excipient compared to the solubility of Drug: phospholipids alone. This might be due to the solubilizing capacity of Ethanol and surface active property of Labrasol and Tween 80. On considering the above results, the formula with Labrasol (60 mg) showed a greater solubility enhancement, when compared to others. So, this formula was used for further studies in formulation development. Hence the ratio corresponding to 30:30:30:60 of Drug: Soy PC: DMPC: Labrasol was selected for further formula optimization.
PROTOTYPE OF FORMULA DEVELOPMENT :
The obtained ratio of 1:1:1:2 of Drug, Soy PC, DMPC and Labrasol showing highest solubility of drug in water was used for the development studies. Unit weight of the liquid formulation equivalent to 60 mg of Raloxifene was filled in to a hard gelatin capsule and its dissolution was carried out in discriminating conditions. The filtered samples were diluted and analyzed using HPLC at 287 nm. A comparison was done with the API and FIONA. Table No.13 and 14, below gives the composition and dissolution profiles of API, FIONA and formulation.
Table No.13: Formula composition of RLX-1

Table No.14: Dissolution results for RLX-1

Fig. No.4: Comparative dissolution profiles of API, FIONA and Lipid based Formulation (RLX-1)

The above results revealed the inefficiency of the RLX-1 formulation to enhance the dissolution profile of the drug when compared to the API and FIONA. Though Labrasol was used as a surfactant, still the dissolution could not be enhanced. So, further studies were carried out by converting the liquid formulation to a solid one using an adsorbent.
CONVERSION INTO A SOLID INTERMEDIATEFOR ENCAPSULATION:
The above liquid formulation was converted into a solid product using an adsorbent. Two adsorbents were screened for the dissolution enhancement of the drug. Both anhydrous Lactose and Aerosil 200 used were of hydrophilic nature. The filtered samples were diluted and analyzed using HPLC at 287 nm and a comparison was done with the API and FIONA. Table No.15 and 16, below gives the composition and dissolution profiles of API, FIONA and formulations.
Table No.15: Formula composition of RLX-2 and RLX-3

Table No.16: Dissolution results for RLX-2 and RLX-3


Fig. No.5: Comparative dissolution profiles of API, FIONA and Lipid based Formulations (RLX-2 and RLX-3)
The above results of the formulation, after converting to a solid product indicated to be useful as there was an improvement in the dissolution profiles compared to that of the liquid formulation alone. Aerosil 200 showed slightly a higher dissolution profile compared to that of Lactose. But the difference was not markedly good. So, further studies were carried out to enhance the dissolution profile and screen the adsorbents.
EFFECT OF PROPYLENE GLYCOL AND TWEEN80:
In these, the drug was initially dissolved individually in propylene glycol and Tween 80 followed by the lipids during the process of formulation to obtain two formulations, RLX-4,RLX-5. The dissolution samples after filtration were diluted and analyzed using HPLC at 287 nm. A comparison was done with the API and the marketed product (FIONA). Table No.17 and 18, below gives the composition and dissolution profiles of FIONA, API and formulations.
Table No.17: Formula composition of RLX-4 and RLX-5
Table No.18: Dissolution results for RLX-4 and RLX-5
Fig. No.6: Comparative dissolution profiles of API, FIONA and Lipid based Formulations (RLX-2 and RLX-3)

The above results indicated the positive effect of Propylene glycol and Tween 80 in the formulation. Though Propylene glycol enhanced the dissolution profile but it could not overcome the FIONA’s dissolution profile. But Tween 80 was able to enhance the dissolution profile to a better extent compared to FIONA. Hence in further studies the combined effect was investigated.
COMBINED EFFECT OF PROPYLENE GLYCOL AND TWEEN 80:
For this a formulation was containing Propylene glycol as a solvent and Tween 80 was prepared and its dissolution profile was checked in the discriminating conditions using HPLC at 287 nm. A comparison was done with the API and the marketed product (FIONA). Table No.19 and 20, below gives the composition and dissolution profiles of FIONA, API and formulation.
Table No.19: Formula composition of RLX-6

Table No.20: Dissolution results for RLX-6

Fig. No.7: Comparative dissolution profiles of API, FIONA and Lipid based Formulation (RLX-6)

Based on the above data, the positive effect of Propylene glycol and Tween 80 on the dissolution rate of the formulation could be estimated. Propylene glycol and Tween 80 when used individually could also enhance the dissolution profile but when used combiningly, the enhancement was higher i.e almost double the dissolution profile of the FIONA. This might be due to the combined effect of Propylene glycol as a solvent, Labrasol as a surfactant and Tween 80 as a co-surfactant. Hence, further optimization of the formula was carried out with this formula (RLX-6) including Drug, Soy PC, DMPC, Labrasol, Propylene glycol and Tween 80.
OPTIMIZATION OF FORMULA:
The formula (RLX-6) containing drug, phospholipids, Labrasol, Propylene glycol and Tween 80 showed an enhanced dissolution profile when compared to the FIONA. But the RLX-6 formula unit weight was around 1000 mg, which was very high. In order to reduce the fill weight the concentrations of the Labrasol, Propylene glycol, Tween 80 and Aerosil 200 were reduced or altered. Table No.21 and 22, below gives the composition and dissolution profiles of FIONA, API and formulations.
Table No.21: Formula composition of RLX-7

Table No.22: Dissolution results for RLX-7
The dissolution profile was superior for the formulation when compared to FIONA. Hence this formulation can concluded as the optimized one. Further the liquid formulation of RLX-7 i.e. without the adsorbent was formulated as RLX-8 to investigate the dissolution profile of optimized liquid formulation. A comparative study of dissolution profile for liquid (RLX-8) and solid (RLX-7) form of the optimized formula was done. Figure No.8 gives the comparative dissolution profiles of the optimized liquid and solid formulations. Table No.23 and 24, below gives the composition and dissolution profiles of FIONA, API and formulation.
Table No.23: Formula composition of RLX-8

Table No.24: Dissolution results for RLX-8

Fig. No.8: Comparative dissolution profiles of API, FIONA and Optimized Formulations (RLX-7 and RLX-8)

The above results indicated convincing for the formulations RLX-7 and RLX-8 on comparison with API and FIONA. The dissolution profile of RLX-7 (solid form) was more when compared to RLX-8 (liquid form). This might be because of the hydrophilic nature of Aerosil 200 which helped in increased solubility of the drug in water. Hence, RLX-7 was concluded as the optimized formula for Raloxifene HCl Lipid based Formulation. The optimized concentration of ingredients for the lipid based formulation is mentioned below in Table No.25.
Table No 25: Optimized Formula for Raloxifene Hydrochloride Lipid based Formulation

EVALUATION OF FORMULATION:
EVALUATION OF BULK GRANULES:
FLOW PROPERTIES:
The granules (lipid based solid intermediate) were evaluated for its flow properties. Their bulk and tapped densities were estimated and the angle of repose, compressibility index and Hausner ratio were calculated. Table No.26 below gives the data for the flow properties of the granules.
Table No.26: Data for the flow properties of the granules

From the above data, the flow properties of the prepared granules (lipid based solid intermediate) exhibited convincing results. Hence, the granulated lipid formulations had good flow properties.
SOLID STATE CHARACTARIZATION:
The solid state characterization studies were carried out for API, Placebo Lipid formulation and Granulated Lipid formulation.
Table No.27: Sample compositions for Solid State Characterization

DIFFERNTIAL SCANNING CALORIMETRY :
Figure No.9 represents the thermograms of pure API, Placebo Lipid Formulation and Granulated Lipid Formulation. The DSC curve of pure Raloxifene HCl exhibited a single endothermic peak at 267.58º C corresponding to the melting of the drug and the sharp peak indicated its crystallinity. The DSC curve of Granulated Lipid Formulation exhibited three broad endotherms at 75.30º, 145.49º C and 219.02º C corresponding to Labrasol, Soy PC and DMPC respectively, but the drug’s peak was no longer observed. It could be attributed to the destruction of crystal lattice, because of progressive amorphization (or) dissolution into the carriers (or) complete entrapment of the drug in the lipids. Solid state studies did not indicate chemical decomposition of the components (drug and excipients) showing compatibility and formation of homogenous systems.
Fig No 9: Thermogram of API , Placebo , and Granulating Lipid Formation

X – RAY DIFFACTOMETRY :
The X-ray diffraction patterns of the pure dug exhibited its characteristic diffraction peaks at various diffraction angles indicating the crystallinity. In the placebo lipid formulation no characteristic peaks were observed due to the absence of crystallinity in the excipients. But in the granulated lipid formulation reduction and absence of major drug diffraction peaks indicated the presence of drug mostly in amorphous form or completely entrapped within the lipid vesicles. Diffraction patterns of API, Placebo lipid formulation and Granulated lipid formulation were represented in the Figure No.10.
FIG No 10: x – Ray Diffraction patterns of API , Placebo , and Granulated lipid formulation

SCANNING ELECTRON MICROSCPY (SEM)
The surface morphology of the particles can be well investigated using the Scanning electron microscopy. These SEM studies gave the particle shape along with the morphology of the particles. In the Granulated lipid formulation the structures completely with spherical shape vesicles adsorbed with a solid adsorbent. No signs of presence of the crystalline drug particles in the granulated formulation were observed indicating the complete entrapment of the drug particles within the lipid vesicles.
Figure No. 11,12 and 13, shows the SEM images of API, Placebo and Granulated lipid formulation.
Fig. No.11: SEM image of API

Fig. No.12: SEM image of Placebo Lipid Formulation

Fig. No.13: SEM image of Granulated Lipid Formulation

EVALUATION OF CAPSULE DOSAGE FORM
WEIGHT VARIATION TEST:
Weight variation was carried out with 20 tablets and % weight variation along with average weight ± SD was reported. The results were within the USP limits
DISINTEGRATION TEST:
Disintegration test was done to 6 capsules of every formulation and its average was reported.
Percent Drug content:
4 capsules of each batch were used for the estimation of % drug content
Table No 28 : Data for the evaluation of the capsule dosage form
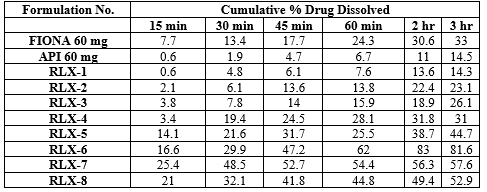
DISSOLUTION :
The dissolution of all the formulations was carried in discriminating dissolution conditions. Along with the dissolution samples the analysis of Blank (diluent), water and placebo were also analyzed using HPLC.
Table No.29: Compiled dissolution data of Raloxifene HCl Lipid based Formulations
DISSOLUTION ACROSS PHYSIOLOGICAL PH BUFFERS :
The optimized formulation (RLX-7) was used further to investigate it’s dissolution across physiological pH buffers and was compared with FIONA. For this the dissolution profiles were investigated in water, USP medium and across pH buffers and finally a 100 % release medium for the formulation was developed for the optimized formulation.
DISSOLUTION STUDIES IN WATER:
The dissolution profile of the optimized formulation was carried out in water (900 mL) with sink conditions being maintained. A comparative study was done considering the FIONA and the formulation. Table No. 30 and 31, corresponds to the dissolution conditions and results carried out in water
Table No 30 : Dissolution conditions in water

Fig No 14 comparative dissolution profiles in water

The above data gave convincing results for the formulation showing a better release profile than that of FIONA, but, 100 % drug release was not observed in either.
DISSOLUTION STUDIES IN OFFICIAL MEDIUM (USP) :
Since the results in water were not promising, the dissolution profiles in the official medium recommended by USP were investigated. The dissolution was carried out in the 0.1 % Polysorbate 80 in 1000 mL water and a comparison was done between the three. Table No.32 and 33 corresponds to the dissolution conditions and results carried out in USP Medium
Table No 32: Dissolution conditions in USP Medium

Table No 33 : Dissolution data in usp medium
Fig No 15: comparative dissolution profiles in USP Medium

This medium also gave enhanced dissolution profile for the formulation when compared to that of FIONA. Though the dissolution profile of the formulation was greater in the USP medium, but 100 % dissolution was not achieved in this medium too. Hence further studies were carried out across physiological buffers.
DISSOLUTION STUDIESACROSS PH BUFFERS:
Since the results in both water and USP medium were not promising in achieving 100 % release, the dissolution profile was investigated across pH buffers. Buffer’s pHs ranging from 1.2 to 7.4 were used for the study. The volumes of the buffers were 900 mL each and a paddle rotation was set to 50 rpm. A comparison was done between the FIONA and formulation. Table No.34, 35, and 36, corresponds to the dissolution conditions and results carried out across pH buffers for FIONA and optimized formulation. A comparative dissolution profiles between the FIONA and Formulation across pH buffers were depicted in figure No.s 16, 17, 18, 19 and 20.
Table No.34: Dissolution conditions in pH buffers

Table No.35: Dissolution data for FIONA across pH

Table No.36: Dissolution data for the Formulation across pH

Fig. No.16: Comparative dissolution profiles in pH-1.2

Fig. No.17: Comparative dissolution profiles in pH-3.0 (50 rpm)

Fig. No.18: Comparative dissolution profiles in pH-4.5

Fig. No.19: Comparative dissolution profiles in pH-6.8

Fig. No.20: Comparative dissolution profiles in pH-7.4

Considering the above results, the formulation showed enhanced dissolution profiles in all of the physiological pH buffers. A sufficient enhancement of the drug’s dissolution could be achieved with the Lipid based formulation when compared to that of FIONA. Hence this lipid based formulation was able to enhance the drug’ solubility and leading to enhanced dissolution velocity. Of these pH buffers, 0.001 N HCl (pH-3.0) showed greater release profile of drug than that of other buffers. So 0.001 N HCl (pH-3.0) was further investigated for the 100 % release conditions. So, in order to get 100 % release rate for the formulation, the dissolution was carried out in 0.001 N HCl (pH- 3.0) with increased paddle rotation from 50 to 75 rpm. A comparative study was done based on the results. Table No.37 and 38, corresponds to the dissolution conditions and results carried out in pH-3.0 buffer at 75 rpm. The most often recognized motor symptoms of PD include bradykinesia, rest tremor, stiffness, and loss of postural reflexes.
Table No.37: Dissolution conditions in pH-3.0 buffer (75 rpm)

Table No.38: Dissolution data in pH-3.0 (75 rpm)

Fig. No.21: Comparative dissolution profiles in pH-3.0 (75 rpm)

Thus, considering the 100 % release rate for the formulation compared to FIONA, this 0.001 N HCl (pH-3.0) with 75 rpm was concluded as the 100 % release medium for the Raloxifene Hydrochloride Lipid based formulation. Table No.39 below gives the optimized dissolution conditions for 100 % release of the Raloxifene HCl lipid based formulation. Table No.40 gives the Comparative dissolution data in 0.001N HCl (pH-3.0) at 50 and 75 rpm.
Table No.39: Optimized dissolution conditions for Raloxifene hydrochloride lipid based formulation

Table No.40: Comparative dissolution data in 0.001N HCl (pH-3.0) at 50 and 75 rpm

Fig. No.22: Comparative dissolution profiles in 0.001N HCl (pH-3.0) at 50 and 75 rpm

STABILITY STUDIES :
A two months storage stability study was conducted on the lipid based formulation. Dissolution runs were conducted on the stressed samples at 15 days interval to assess any changes in release behavior of raloxifene HCl. Accordingly, the formulations were packed in HDPE bottles and charged for stability studies at 25º C / 60 % RH and 40º C / 75 % RH for a period of 2 months. At pre-determined intervals of 15, 30 45 and 60 days, the samples were analyzed for the percent drug content and the drug dissolution rates. The dissolution studies were carried out in the developed 100 % release medium. Table No.41, 42 and 43 corresponds to the dissolution conditions and results carried out for stability studies.
Table No.41: Dissolution conditions for stability studies
Table No 42 : stability results for FIONA

Table No 43 : stability results for Raloxifene HCL Lipid based formulation

Fig. No. 23: Comparative stability data

The above results indicated the intermediate stability of the formulation. As the storage time period increased, there was a slight decrease in the release of Raloxifene HCl from the formulation. A decrease of around 4 % at 25°C / 60% RH and around 6 % at 40°C / 75% RH in the dissolution rate of the drug was observed in the formulation. While the marketed formulation showed good results at both temperature conditions. The decrease in stability as a result of decreasing dissolution rates in the formulation could be attributed to the precipitation of drug from the solvent or formation of crystalline mass upon storage for extended periods. However addition of appropriate stabilizers (e.g., polyvinyl alcohols) could potentially increase the storage stability of the formulation for longer periods of time.
SUMMARY AND CONCLUSION
The goal of any drug delivery system is to provide a therapeutic amount of drug to the proper site in the body and also to achieve and maintain the desired plasma concentration of the drug for a particular period of time. However, low water solubility, incomplete release of the drug, shorter residence times of dosage forms in the upper GIT leads to lower oral bioavailability. Such limitations of the conventional dosage forms have paved way to an era of novel drug delivery systems.Raloxifene hydrochloride is a poorly water soluble drug having an oral bioavailability of only 2%. This is due to the first pass metabolism by the intestinal UGT’s which lead to high inter subject variability. Raloxifene HCl belongs to the category of Selective Estrogen Receptor Modulator (SERM), used in the treatment of Osteoporosis and prevention of invasive breast cancer in post-menopausal women. Thus in order to overcome these drawbacks of the drug molecule, a lipid based formulation approach was selected. For this initially, the solubility studies across pH and water indicated the highest solubility of raloxifene HCl in pH-3.0 (0.001 N HCl). Lipid excipients were screened in order to enhance the solubility of Raloxifene HCl in water. Based on these results Soy PC, DMPC and Labrasol® were selected as lipid ingredients. Using the above excipients and along with a solvent and co-surfactant, lipid based formulations of Raloxifene HCl were prepared using the conventional methods of preparation and without using any organic solvent during the process and finally converting into a solid intermediate for capsule fillling. The prepared formulations were evaluated in discriminating dissolution medium for the dissolution profile. And optimized formulation was screened by comparing with that of the API and marketed product (FIONA). The prepared formulations were characterized for their powder flow characteristics along with the Physical characterization like DSC, XRD and SEM studies. The optimized formulation showed fairly acceptable values for all the parameters evaluated. The physical characterization revealed the amorphous nature or decrease in the crystallinity of the drug molecule while present in the optimized formulation. The dissolution profiles in the fed and fasted state simulated intestinal fluid showed a very insignificant effect or almost negligible effect of food on the dissolution rate of Raloxifene HCl from the lipid based formulation. The optimized lipid based formulations was charged to accelerated stability studies for a period of two months. But there was a reduction in the release rate of Raloxifene HCl from the lipid based formulation showing intermediate stability, which commands for the use of stabilizers in the formulation for attaining significant stability. From the in vitro dissolution and solid state characterization data it is apparent that, the presence of Soy PC, DMPC and Labrasol® together as solubilizers and carriers has a significant impact on dissolution characteristics.
Thus the prepared lipid based formulation proved to be a potential technology for enhancing the transfer of poorly water soluble lipophilic compounds to the aqueous phase, thus enhancing the bioavailability.
ACKNOWLEDGEMENT :
We would like to express our sincere gratitude, who supported for the completion of this research article and Technical support for this study was provided by faculty from the Department of Pharmaceutics. The authors wish to thank Dr.SD. Jilani. and Dr.K.Vinodkumar for their work in authenticating plant samples. Thanks to V.Satyasri, M.Hyma for their suggestion on preparing the manuscript.
CONFLICT OF INTEREST :
No conflict of interest.
ABBREVATIONS

REFRENCES
Wilson, C.G., Mahony, B.O., 1997. The behavior of fats and oils in the upper G. I. Tract. Bulletin Technique Gattefosse. 90, 13–18.
Wilson, C.G., Mahony, B.O., 1997. The behavior of fats and oils in the upper G. I. Tract. Bulletin Technique Gattefosse. 90, 13–18.


Thota Srinivas, Nilanjana Das, Kondaveeti Vyshnavi, Ponduri Gowtham, Veena Navya Chandana, A. Jeevan Kumar, Jambil MD, Formulation & Development Of Capsular Device Incorporated With Granulated Lipid Formulation Of Raloxifene Hydrochloride, Int. J. of Pharm. Sci., 2024, Vol 2, Issue 6, 49-78. https://doi.org/10.5281/zenodo.11440569
 10.5281/zenodo.11440569
10.5281/zenodo.11440569
