Department of Pharmaceutics SDMVMS SVP College of Pharmacy Hatta Dist.- Hingoli Maharashtra.
The present study was aimed at the development and evaluation of an in situ nasal gel formulation of Montelukast Sodium Six formulations (F1–F6) of Montelukast Sodium in situ nasal gel were prepared using varying concentrations of chitosan and Carbopol 934 to optimize gel strength, viscosity, Mucoadhesion, and drug release. All formulations were subjected to comprehensive evaluation. pH of All batches were within the range of 5.7–5.9, suitable for nasal application. Drug Content study showed high uniformity was observed (98.3–99.5%), with F3 showing the highest content (99.5 ± 0.8%).Viscosity was found in ranged from 150 cps (F1) to 290 cps (F6); F3 (210 cps) provided the most balanced flow properties. Gelling Capacity: F3 and F4 showed +++ (instant and strong gelation), making them suitable for rapid in situ transformation upon nasal administration. Gel Strength: F3 exhibited adequate strength (38.9 ± 1.7 sec) to retain the formulation at the absorption site. Mucoadhesive Strength showed that increased with polymer content. F3 showed a favorable value (1382 ± 50 dyne/cm²), promoting better retention in the nasal cavity. These results collectively established F3 as the optimized formulation, offering ideal clarity, pH, drug content, gelling behavior, and mucoadhesion. The in vitro diffusion study using a Franz diffusion cell with goat nasal mucosa demonstrated varying drug release profiles; F1 released 98.24 ± 1.34% in 5 hours due to lower viscosity. F6 showed the slowest release (76.92 ± 1.88% at 6 hours) due to a dense polymeric network. F3 demonstrated controlled release the optimized formulation F3 was evaluated for 3-month stability under ICH conditions. The results confirmed that the formulation retained physical and chemical stability over time and is suitable for long-term storage and usage.
The nasal drug delivery received a great deal of attention for its convenient, promising, and reliable way of systemic administration for drugs, especially for those drugs which are ineffective orally and those which must be administered by injections. This route provides a large surface area, porous endothelial membrane, high total blood flow, bypassing the first-pass metabolism, and ready accessibility. Furthermore, nasal mucosa is permeable to more compounds than the gastrointestinal tract due to the absence of pancreatic, gastric enzymatic activities, and interference by gastrointestinal contents. The early recorded historical application of nasal drug delivery was restricted to topical applications of drugs intended for only local effects. However, in recent times, its application grown to include a wide range of targeted areas in the body to produce local and systemic effects. Nasal drug delivery also finds a special place in the traditional system of medicine such as the Ayurvedic system of Indian medicine which is called as “Nasya karma” and is a well-recognized way of treatment.1 In therapeutics, nose forms an important part of the body for faster and higher level of drug absorption with the possibility of self-administration. Drugs are ranging from small micromolecules to large macromolecules such as peptide/proteins, hormones, and vaccines, are being delivered through the nasal cavity. It is reported that lipophilic drugs are generally well absorbed from the nasal cavity with pharmacokinetic profiles often identical to those obtained following an intravenous injection with a bioavailability approaching up to 100% in many cases. Large absorption surface area and high vascularization lead to fast absorption. In emergency, nasal route can be used as a substitute route of parenteral administration. Drugs are rapidly absorbed from the nasal cavity after intranasal administration, resulting in rapid systemic drug absorption. An approach if made for increasing the residence time of drug formulations in the nasal cavity can result in improved nasal drug absorption. Depending on the desired site of drug action, the drug to be inhaled needs to be adjusted to particle size, concentration, and chemical form to ensure a local or systemic drug action.1, 2,3. In Latin, in situ means ‘in position’ or ‘in its original place’.For the past 30 years, greater attention has been directed towards the development of controlled and sustained drug delivery systems. A vast amount of research has been carried out in designing polymeric systems such as in situ gels. In situ gel formation of drug delivery systems can be defined as a liquid Formulation generating a solid or semisolid depot after administration. In situ activated gel, forming Systems are those which are when exposed to physiological conditions that will shift to a gel phase. This new concept of manufacturing a gel insitu was suggested for the first time in the early 1980s. Gelation occurs via the cross-linking of polymer chains that can be achieved by covalent bond formation (chemical cross-linking) or non-covalent bond formation (physical cross-linking). The routes of administration for in situ gel could be oral, ocular, rectal, vaginal, injectable and intra-peritoneal. “Gel”is that the state which exists in between liquid and solid, which consists of physically crosslinked networks of long polymer molecules, with liquid molecules trapped within three- dimensional polymeric network swollen by a solvent. This system is a liquid aqueous solution before the administration and a gel at physiological conditions. Prolonged and sustained release of the drug is reproducible, and the insitu gel is biocompatible, with magnificent stability and reliable quantities of medication, making it more accurate. There are various routes for in situ gel drug delivery, for example, oral, ocular, vaginal, rectal, intravenous, intraperitoneal, etc. In situ gel is a new dosage form that has been applied in nasal drug delivery recently. Compared with other liquid nasal formulations, nasal in situ gels are administered as low viscosity solutions into the nasal cavity, and upon contact with the nasal mucosa, the polymer changes conformation producing a gel, so it cannot only prolong the contact time between the drug and also the absorptive sites within the cavity but also release drug slowly and continuously, hence, it's especially useful for those drugs used chronically. The phase transition can be induced by a shift in pH, a shift in temperature or by the presence of cations.4, 5, 6
MATERIALS AND METHODS
MATERIALS-
Montelukast Sodium was obtained as gift sample from Glenmark Pharma, Mumbai, Chitosan & Carbopol was purchased from S. D. Fine Chemicals. All other chemicals and solvents are analytical grade.
METHODS.
Drug Excipients Compatibility Studies.
Drug-excipient compatibility studies are an essential part of preformulation and formulation development processes in the pharmaceutical industry. These studies assess the compatibility of a drug substance with various excipients that are used to formulate the final dosage form. The primary purpose of drug-excipient compatibility studies is to evaluate potential interactions between the drug substance and excipients. These studies aim to identify any chemical, physical, or mechanical interactions that could affect the stability, efficacy, or safety of the final dosage form. By assessing compatibility early in the development process, formulation scientists can make informed decisions regarding excipient selection, formulation design, and process optimization. Compatibility study of drug with the excipients was determined by I.R. Spectroscopy (Shimadzu, Japan). The pellets were prepared at high compaction pressure by using KBr and the ratio of sample to KBr is 1:100. The pellets thus prepared were examined and the spectra of the drug and other ingredients in the formulations were compared with that of the pure drug.8,9
Formulation of Montelukast Sodium Nasal In Situ Gel
The in situ nasal gel formulations of Montelukast Sodium were prepared by the mechanism of pH – triggered system using the cold method under aseptic conditions. Firstly, the required quantity of Chitosan was slowly dispersed in small amount of 1% v/v acetic acid solution with continuous stirring using a magnetic stirrer until a clear, homogeneous solution was obtained. Simultaneously, Carbopol 934 was soaked in a small quantity of distilled water and allowed to hydrate for at least 2 hours, then stirred to form a smooth dispersion. In a separate beaker, Montelukast Sodium was dissolved in distilled water and mixed with solubilizers such as PEG 400 under moderate stirring. After both polymer solutions were ready, the Carbopol dispersion was slowly added to the Chitosan solution with continuous stirring to avoid lump formation. The drug solution was then added gradually to the polymer mixture, ensuring uniform distribution. Preservatives like Benzalkonium Chloride and isotonicity adjusters such as Sodium Chloride were incorporated into the formulation. The volume was adjusted with distilled water, and the final pH of the formulation was carefully adjusted between 5.5 and 5.8 using either 0.1N NaOH or 0.1N HCl depending on the formulation requirement.10,11. The prepared formulation was kept overnight at 4–8°C to ensure complete hydration and clarity. Finally, the in situ nasal gels were stored in sterile nasal containers or vials and evaluated for various physicochemical parameters. The composition of different formulation of Montelukast Sodium Nasal In situ gel is shown in the table 1
Table 1: Composition of Nasal in Situ Gel of Montelukast Sodium
|
Ingredients (% w/v) |
F1 |
F2 |
F3 |
F4 |
F5 |
F6 |
|
Montelukast Sodium |
0.5 |
0.5 |
0.5 |
0.5 |
0.5 |
0.5 |
|
Chitosan |
0.2 |
0.3 |
0.4 |
0.3 |
0.4 |
0.3 |
|
Carbopol 934 |
0.1 |
0.2 |
0.2 |
0.3 |
0.3 |
0.4 |
|
PEG 400 |
1.5 |
1.5 |
2.0 |
2.0 |
2.0 |
2.0 |
|
Benzalkonium Chloride |
0.01 |
0.01 |
0.01 |
0.01 |
0.01 |
0.01 |
|
Sodium Chloride |
0.9 |
0.9 |
0.9 |
0.9 |
0.9 |
0.9 |
|
Distilled Water |
Q.S. to 100 ml |
Q.S. to 100 ml |
Q.S. to 100 ml |
Q.S. to 100 ml |
Q.S. to 100 ml |
Q.S. to 100 ml |
|
Final pH Adjusted To |
5.5 |
5.5 |
5.5 |
5.5 |
5.5 |
5.5 |
Evaluation of In Situ Gel of Montelukast Sodium
pH Measurement
The pH of each formulation was measured using a digital pH meter calibrated with standard buffer solutions of pH 4.0 and 7.0 before use. About 10 ml of the gel solution was taken in a beaker, and the electrode of the pH meter was immersed into the sample. The reading was recorded once it stabilized. The pH was adjusted between 5.5 and 6.0, which is suitable for nasal administration to avoid irritation and ensure comfort during application.12
Drug Content Estimation
To determine the drug content, 1 ml of the formulation was diluted with phosphate buffer (pH 6.4) up to 100 ml in a volumetric flask. The solution was sonicated for 10 minutes to ensure complete solubilization of the drug, followed by filtration using Whatman filter paper No. 41. The resulting solution was analyzed using a UV-Visible spectrophotometer at 287 nm. The absorbance was measured, and the drug content was calculated using a previously constructed standard calibration curve.13
Viscosity Measurement
The viscosity of the formulations was determined using a Brookfield viscometer fitted with an spindle no 62. About 25 ml of each formulation was placed in the sample holder, and the spindle was rotated at 50 rpm at 25 ± 1°C. The viscosity (in centipoise, cps) was recorded after the dial reading stabilized. Measurements were performed in triplicate for accuracy.14
Gelling Capacity
Gelling capacity was evaluated by simulating the nasal environment using simulated nasal fluid (SNF) of pH 6.4. A fixed volume (1 ml) of the formulation was added to 2 ml of SNF in a test tube maintained at 37 ± 0.5°C. The time taken for gel formation and the duration for which the gel remained intact without dissolving or breaking was visually recorded. Gelling capacity was scored as15
Gel Strength
Gel strength was measured using a modified method in which 50 g of the prepared gel (formed by adding formulation to SNF) was placed in a 100 ml graduated cylinder. A weighed piston (20g) was gently placed on top of the gel, and the time (in seconds) required for the piston to sink 5 cm deep into the gel was recorded using a stopwatch. The longer the time, the stronger the gel network formed by the polymers.16
Mucoadhesive Strength
Mucoadhesive strength was measured using a modified physical balance method or tensile detachment method. Freshly excised goat nasal mucosa obtained from local slaughter house was mounted on a glass slide using cyanoacrylate glue. Another mucosal section was attached to the hanging side of the balance. A small amount of gel (about 0.1 g) was placed between the two mucosal surfaces and allowed to adhere for 2 minutes. Water was then added dropwise into the beaker on the other side of the balance until the two mucosal surfaces detached. The weight required to detach the surfaces was converted to dyne/cm² to calculate mucoadhesive strength using the formula17
Force = (m × g) / A, where
m = mass added (g)
g = acceleration due to gravity (980 cm/s²)
A = contact area (cm²)
In-Vitro Drug Diffusion Study
The in vitro drug diffusion study of Montelukast Sodium in situ nasal gel was performed using a Franz diffusion cell apparatus to evaluate the drug permeation through biological membrane. Freshly excised nasal mucosa from a healthy goat was obtained from a local slaughterhouse. The mucosal tissue was carefully separated from the underlying cartilage, washed thoroughly with phosphate-buffered saline (pH 6.6) to remove mucus and blood, and then stored in phosphate buffer at 4°C. Before the experiment, the tissue was equilibrated to room temperature and inspected for uniform thickness and absence of tears. The Franz diffusion cell consisted of a donor and receptor compartment with an effective diffusion area of approximately 1.76 cm². The receptor compartment was filled with phosphate buffer saline (pH 6.4), simulating nasal physiological conditions. The buffer was degassed and maintained at 37 ± 0.5°C using a thermostatically controlled water jacket. A small magnetic bead was placed in the receptor chamber and stirred continuously at 50 rpm to ensure uniform drug distribution in the receptor medium. The excised nasal mucosa was mounted between the donor and receptor compartments with the mucosal side facing the donor compartment. Care was taken to avoid the formation of air bubbles at the interface. A known amount of Montelukast Sodium in situ nasal gel (a dose equivalent to 10 mg of the drug) was accurately weighed and placed on the mucosal surface in the donor compartment. The donor chamber was sealed with parafilm to prevent evaporation and contamination. At specific time intervals (0.5, 1, 2, 3, 4, 5, and 6 hours), 1 ml of sample was withdrawn from the receptor compartment using a micropipette or syringe and immediately replaced with an equal volume of fresh pre-warmed buffer to maintain sink conditions. The withdrawn samples were filtered if necessary and analyzed using a UV-Visible spectrophotometer at the predetermined λmax of Montelukast Sodium (around 287 nm) to determine the amount of drug diffused. The cumulative amount of drug permeated per unit area was calculated and plotted against time to obtain the drug release profile.18,19
Stability Study
The accelerated stability studies were carried out on In situ nasal gel stired in glass vial according to ICH (International Conference on Harmonization) guidelines on optimized formulation. A sufficient quantity of in situ gel in glass vial bottles was placed in stability chamber (Labline India) at 40 ± 2 °C, and the samples were withdrawn at predetermined time interval. Changes in the appearance, drug content, gelling strength, and in vitro drug diffusion of the stored formulations were investigated.20,22
RESULTS AND DISCUSSION
Compatibility Studies (FT-IR)
The IR spectra of a drug-polymer physical mixture was compared to that of a plain drug. Characteristic sharp distinctive peaks seen in pure drug Montelukast Sodium, was compared with the spectra of drug and polymer mixtures. It was observed that characteristic peaks those found on pure drug spectra were still detectable in spectra of drug polymer mixture spectra, showing that the drug and polymer have no interaction. FTIR spectra of pure drug Montelukast Sodium and drug polymer physical mixture was shown in figure 1 and 2 respectively.
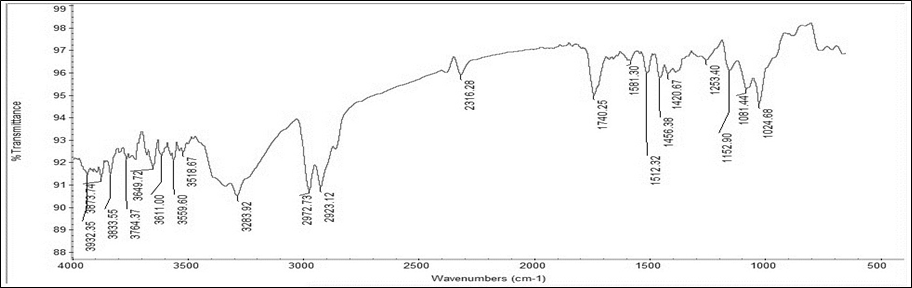
Figure 1 IR spectra of pure drug Montelukast Sodium.
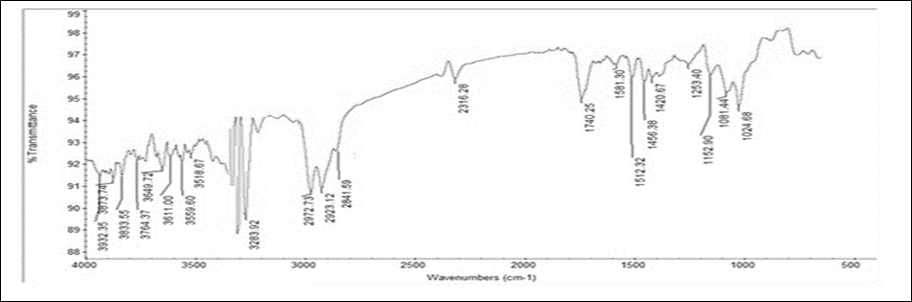
Figure 2 IR Spectra of Optimized Montelukast Sodium with Chitosan.
Evaluation of In Situ Gel of Montelukast Sodium
The in situ nasal gel formulations of Montelukast Sodium (F1–F6) were evaluated for various physicochemical parameters including appearance, pH, drug content, viscosity, gelling capacity, gel strength, and mucoadhesive strength. These evaluations were essential to ensure the safety, stability, and efficacy of the formulation for intranasal administration.
Appearance
The appearance of the in situ nasal gel formulation was assessed visually. Formulations F1 to F3 appeared clear and transparent, which is desirable for nasal formulations to ensure patient acceptability and uniform drug distribution. In contrast, formulations F4 to F6 exhibited a slightly hazy appearance, which could be attributed to higher polymer concentrations leading to reduced clarity. The results are shown in table 2.
pH Measurement
The pH of each formulation was measured using a calibrated digital pH meter. The pH of all formulations ranged from 5.7 to 5.9, which is within the acceptable range for nasal mucosa (pH 4.5–6.5), thereby minimizing the risk of irritation or discomfort upon administration. The optimized formulation F3 showed a pH of 5.8 ± 0.03, indicating good compatibility with the nasal environment. The results are shown in table 7.2.
Drug Content Estimation
The drug content of all formulations was found to be within acceptable limits, ranging from 98.3 ± 1.1% (F1) to 99.5 ± 0.8% (F3), indicating uniform drug distribution throughout the gel matrix. The optimized formulation F3 exhibited the highest drug content, ensuring dose uniformity.
Viscosity Measurement
The viscosity of the formulations was determined using a Brookfield. Viscosity is a critical parameter for nasal gels as it influences the drug release and residence time. A progressive increase in viscosity was observed from F1 (150 ± 5.2 cps) to F6 (290 ± 7.8 cps), due to increased polymer concentration. F3, the optimized formulation, exhibited a balanced viscosity of 210 ± 5.7 cps, which is ideal for ease of administration while ensuring adequate retention at the site of absorption.
Gelling Capacity
Gelling capacity was evaluated by simulating the nasal environment using simulated nasal fluid (SNF) of pH 6.4. The gelling capacity of the formulations varied significantly. F1 showed weak gelation (+), F2 exhibited slow gel formation (++), whereas F3 and F4 demonstrated instant and strong gelation (+++), which is considered ideal for in situ gelling systems. The strong gelling behavior of F3 ensures rapid transformation into gel upon nasal administration, enhancing drug retention and absorption.
Gel Strength
Gel strength, which indicates the mechanical strength of the formed gel, ranged from 25.6 ± 1.4 seconds (F1) to 50.2 ± 2.0 seconds (F6). The optimized formulation F3 showed a gel strength of 38.9 ± 1.7 seconds, which is adequate to maintain structural integrity in the nasal cavity without causing discomfort. Mucoadhesive Strength Mucoadhesive strength was measured using a modified physical balance method. The mucoadhesive strength is a vital parameter to assess the formulation's ability to adhere to the nasal mucosa, thus prolonging residence time and enhancing drug absorption. It was found that F3 had a mucoadhesive strength of 1382 ± 50 dyne/cm², which was significantly higher than F1 and F2 and suitable for effective nasal delivery. Further increase in mucoadhesive strength in F4 to F6 (up to 1583 ± 60 dyne/cm²) could be due to higher polymer content, although too strong adhesion may affect patient comfort. Overall, from the above evaluation parameters it was concluded that, formulation F3 exhibited an ideal balance of clarity, appropriate pH, and high drug content, optimal viscosity, instant gelling behavior, suitable gel strength, and sufficient mucoadhesive strength, making it the optimized formulation among all batches tested.
Table 2: Evaluation Data of Montelukast in Situ Nasal Gel Formulations (F1–F6)
|
Parameter |
F1 |
F2 |
F3 |
F4 |
F5 |
F6 |
|
Appearance |
Clear, transparent |
Clear, transparent |
Clear, transparent |
Slightly hazy |
Slightly hazy |
Slightly hazy |
|
pH |
5.8 ± 0.04 |
5.9 ± 0.05 |
5.8 ± 0.03 |
5.9 ± 0.04 |
5.7 ± 0.05 |
5.8 ± 0.05 |
|
Drug Content (%) |
98.3 ± 1.1 |
99.1 ± 1.0 |
99.5 ± 0.8 |
98.9 ± 1.2 |
98.4 ± 1.3 |
99.0 ± 1.1 |
|
Viscosity (cps) |
150 ± 5.2 |
185 ± 6.1 |
210 ± 5.7 |
240 ± 6.3 |
275 ± 7.2 |
290 ± 7.8 |
|
Gelling Capacity |
+ |
++ |
+++ |
+++ |
++ |
++ |
|
Gel Strength (sec) |
25.6 ± 1.4 |
32.4 ± 1.6 |
38.9 ± 1.7 |
42.3 ± 1.8 |
48.6 ± 1.9 |
50.2 ± 2.0 |
|
Mucoadhesive Strength (dyne/cm²) |
1124 ± 45 |
1240 ± 48 |
1382 ± 50 |
1448 ± 53 |
1525 ± 56 |
1583 ± 60 |
In-Vitro Drug Diffusion Study
The in vitro drug diffusion profile of Montelukast Sodium nasal in situ gel formulations (F1–F6) was evaluated using Franz diffusion cells across goat nasal mucosa. The study was carried out over a 6-hour period, and the cumulative percentage drug release was recorded at predetermined intervals. The results demonstrated significant variations in the drug release rates among different formulations, primarily influenced by the polymer concentration and viscosity. Formulation F1 showed the fastest release, with 98.24 ± 1.34% drug release within 5 hours, followed by F2 with 95.63 ± 1.51%, indicating their comparatively lower viscosity and weaker gel matrix, which allowed faster drug diffusion. In contrast, F6 showed the slowest release, with only 76.92 ± 1.88% drug diffused at the end of 6 hours. This may be attributed to its higher polymer content, which results in a more viscous and stronger gel structure, restricting the diffusion of the drug. The optimized batch F3 displayed a controlled and sustained drug release, reaching 95.24 ± 1.64% at the end of 6 hours. The gradual and extended release from F3 is attributed to its balanced polymer concentration, which provides an ideal gel consistency, adequate mucoadhesiveness, and effective diffusion control. The gel formed by F3 provided a stable matrix, allowing a sustained diffusion of Montelukast Sodium, aligning well with the desired nasal delivery profile. Formulations F4 and F5 also showed moderately sustained release profiles with87.22 ± 1.35% and 79.88 ± 1.32% drug release respectively at 6 hours, which again correlates with their gelling capacity and viscosity. Their comparatively slower release than F3 suggests a higher polymer concentration leading to a more entangled network structure, further slowing down the drug diffusion. Overall, the results suggest that polymer concentration plays a critical role in modulating the diffusion rate. The optimized formulation F3 achieved a desirable balance between rapid gelation, adequate gel strength, and sustained drug release, making it the most suitable candidate for effective intranasal delivery of Montelukast Sodium. Data for in vitro drug release of montelukast sodium was shown in figure 3.
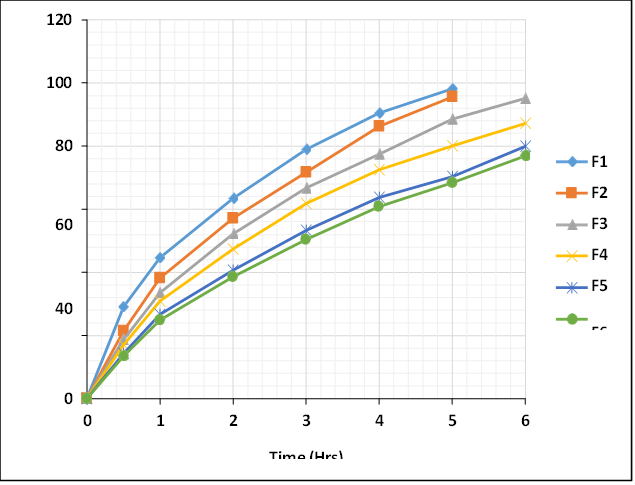
Figure 3: Comparative In vitro Drug Diffusion Profile of Formulation F1 to F6
Stability Study
The optimized in situ nasal gel formulation F3 was subjected to stability testing under standard conditions (typically 25°C ± 2°C / 60% RH ± 5% RH) for a period of three months, to assess the physical integrity, drug content, gel strength, and drug release behavior over time. After 3 months of storage, the appearance of the formulation remained clear and transparent, indicating no visible signs of physical degradation such as precipitation, turbidity, or phase separation. This demonstrates that the formulation maintained its physical stability under the tested storage conditions. The drug content slightly decreased from 99.5 ± 0.8% to 98.9 ± 0.6%, which remains well within the acceptable range (typically 90–110%). This minor reduction is likely due to natural degradation over time and confirms the chemical stability of Montelukast Sodium in the gel matrix.The gel strength showed a slight decrease from 38.9 ± 1.7 seconds to 36.5 ± 1.5 seconds, which indicates that the gelling ability of the formulation was well preserved over time. The slight reduction may be due to minor structural changes in the polymer network. The in vitro drug release after 3 months showed a minimal reduction from 95.24 ± 1.64% to 95.02 ± 1.21%, indicating that the drug diffusion characteristics were largely preserved. The negligible change suggests that the formulation maintained its controlled release behavior even after prolonged storage.The result concluded that, the optimized formulation F3 remained physically and chemically stable over the 3-month period. The negligible changes in drug content, gel strength, and drug release profile demonstrate that the formulation can be stored under ambient conditions without compromising its performance, thereby confirming its suitability for long-term use and commercialization. The results of stability data were shown in table 3.
Table 3: Stability Data of Optimized In situ Nasal gel Formulation F3
|
Formulation Code |
Parameter |
Before storage (0 month) |
After storage (3 month) |
|
F3 |
Appearance |
Clear, transparent |
Clear, transparent |
|
Drug Content (%) |
99.5 ± 0.8 |
98.9±0.6 |
|
|
Gel Strength (Sec) |
38.9 ± 1.7 |
36.5±1.5 |
|
|
% Drug Release |
95.24±1.64 |
95.02±1.21 |
CONCLUSION
The study successfully developed and optimized a Montelukast Sodium in situ nasal gel formulation using natural and synthetic polymers to enhance therapeutic efficacy through sustained intranasal delivery. Among six formulations, F3 emerged as the optimized batch, offering a clear, stable, pH-compatible, mucoadhesive gel with excellent gelling behavior and sustained drug release.The formulation followed zero-order kinetics with a Fickian diffusion mechanism, ensuring predictable and extended drug availability. Furthermore, stability studies validated the long-term effectiveness and robustness of the optimized gel. This novel nasal in situ gel system holds promise as a patient-friendly, efficient alternative to conventional oral or inhalation routes for Montelukast Sodium, particularly for the management of allergic conditions requiring rapid and sustained relief.
REFERENCES

Piyusha Rathod, Umesh Jadhao*, G. N. Dhembre, S. A. Wathore, S. T. Thoke, D. A. Rathod, Formulation and Evaluation of Nasal In-Situ Gel of Montelukast Sodium, Int. J. of Pharm. Sci., 2025, Vol 3, Issue 8, 1341-1351. https://doi.org/10.5281/zenodo.16848374
 10.5281/zenodo.16848374
10.5281/zenodo.16848374
