
We use cookies to ensure our website works properly and to personalise your experience. Cookies policy
Swami Vivekanand college of pharmacy Udgir, Maharashtra- 413517.
This research focuses on the creation and assessment of immediate release tablets containing Nateglinide, a medication used to treat Type 2 diabetes mellitus. These formulations are designed to dissolve quickly, ensuring rapid drug release, which enhances patient adherence and therapeutic effectiveness. Nateglinide tablets were produced using the wet granulation method, incorporating various excipients, such as starch, lactose, and sodium starch glycolate. A total of six formulations (F1–F6) were developed and assessed for both pre-compression and post-compression characteristics, including angle of repose, bulk density, hardness, friability, thickness, weight variation, disintegration time, and in vitro drug release. Among these, formulation F2 demonstrated the most advantageous properties, with a rapid disintegration time of 13.3 min and the highest cumulative drug release of 87.16% within 60 min. The compatibility between the drug and excipients was verified using FTIR and DSC analyses. Stability tests showed no significant changes in drug release over the month. These findings indicate that the optimized formulation is a promising strategy for achieving immediate therapeutic effects with nateglinide.
Immediate Release:
The oral route is the most popular for systemic effects because of its ease of ingestion, pain avoidance, versatility, and most importantly, patient compliance. Solid oral delivery systems (especially tablets) are the preferred choice among all drug delivery systems because they do not require special treatment and are therefore less expensive to manufacture. Likewise, immediate-release tablets were more acceptable than the other types. Based on their drug-release characteristics, tablets can be classified into three types: immediate, extended, and delayed release. For immediate-release tablets, the drug is intended to be released rapidly after administration, or the tablet is dissolved and administered as a solution. This is the most common type of tablet and includes disintegrating, chewable, effervescent, sublingual, and buccal tablets. Immediate release (Immediate) tablets are designed to disintegrate and release their medication with no special rate-controlling features, such as special coatings and other techniques. In the pharmaceutical industry, manufacturers of generic tablets usually focus on optimizing the excipient mixture composition to obtain a product that meets the established standards. Immediate-release tablets are designed to disintegrate and release their dosage form without special rate-controlling features, such as special coatings and other techniques. Immediate-release tablets disintegrate swiftly and dissolve to release the medicaments. The oral bioavailability of a drug depends on disintegration, dissolution, and various physiological factors. An immediate-release dosage form helps a manufacturer diversify the market while simultaneously offering patients a convenient dosage form or regimen.
Desired Criteria for Immediate-Release Drug Delivery System:
? Immediate release dosage form should-
?In the case of solid dosage forms, they should dissolve or disintegrate in the stomach within a short period.
? Have a pleasing mouth feel.
It should leave minimal or no residue in the mouth after oral administration.
? Exhibits low sensitivity to environmental conditions such as humidity and temperature.
? Rapid dissolution and absorption of the drug, which may produce a rapid onset of action.
Ideal Properties
Immediate release dosage form should
1. In the case of solid dosage forms, it should dissolve or disintegrate in the stomach within a short period.
2. It should show the first absorption and dissolution of the drug.
ADVANTAGES
An immediate release pharmaceutical preparation offers
1. Improved stability and bioavailability.
2. Decreased disintegration and dissolution times for immediate-release oral dosage forms.
3. Suitable for controlled, sustained release of actives.
4. A high drug loading is possible.
5. Ability to provide the advantages of liquid medication in the form of a solid preparation.
DISADVANTAGE
1. Frequent dosing is necessary for drugs with a short half-life.
2. Drug release at a time may produce high plasma concentrations, which may result in toxicity.
The immediate-release tablets offer several advantages, including improved stability, bioavailability, and decreased disintegration and dissolution times. These formulations allow for high drug loading and provide the benefits of liquid medication in a solid form. However, they also have drawbacks, such as the need for frequent dosing for drugs with short half-lives and the potential for high plasma concentrations that may lead to toxicity.
Literature Review
1. Monica RP Rao: The development of immodiate release tablet formulations is based on the use of super disintegrants separately or in combination. Seven formulations were prepared using simplex centroid mixture design where sodium starch glycolate, cross carmellose sodium and pre-gelatinised starch were selected as independent variables and dependent variables. Response surface plots were drawn. and optimum formulations were selected by grid search method, Formulations when used individually gave satisfactory results but when used in combination gave better results. The results showed a good relationship between the experimental and predicted values, which confirms the predictability of the model. [1]
2 Nyol Sandeep et al: Immediate drug release dosage forms disintegrate rapidly after administration with enhanced rate of dissolution. The basic approach used in development of tablets is the use of superdisintegrants like Sodium starch glycolate (Primogel, Explotab). These superdisintegrants provide instantaneous disintegration of tablet after administration in stomach. In this field immediate release liquid dosage forms and parenteral dosage form have also been introduced for treating patients. In liquid dosage form can be suspensions with typical dispersion agents like hydroxypropyl methylcellulose, AOT etc. The development of immediate release therapy also provides an opportunity for a line extension in the marketplace, a wide range of drugs e.g., neuroleptics, cardiovascular drugs, analgesics, antihistamines and other drugs can be considered candidates for this dosage form. As a drug entity nears the end of its patent life, it is common for pharmaceutical manufacturers to develop a given drug entity in a new and improved dosage form. A new dosage form allows a manufacturer to extend market exclusivity, while offering its patient population a more convenient dosage form or dosing regimen.
3. Yunxia B et.al: Tablets were prepared by direct compression technique. The granules were evaluated for angle of repose, bulk density, tapped density, bulkiness, compressibility index and hausners ratio. The tablets were evaluated for hardness, thickness, uniformity of weight, friability, wetting time, water absorption ratio, disintegeration time and drug content. In vitro release studies were performed using USPII (paddle method) in 900ml of pH 1.2 at 50 rpm. The physical properties of the prepared tablets did not show any significant varitions and were found to have good physical integrity. Tablets prepared with pharmaburst B2 and carmellose sodium showed a lesser disintegerationtime and wetting time of 27.40.10 and 3840.13 seconds respectively.
4. Paul J Hesketh el at: New sight into the pathophysiology of chemotheraphy-induced nausea and vomiting, a better understanding of patient at risk, and the availability of new antiemetic agents have all contributed to substantial improvement in emetic control. This review focuses on our current understanding of chemotherapy induced nausea and vomiting and the status of pharmacologic interventions in its prevention and treatment.
5. Molassiotis, A. et al: He evaluated acute chemotherapy-induced nausea and vomiting (CINV) prophylaxis; provided sufficient information to permit determination of the emetogenicity of the antineoplastic therapy administered or the study investigators stated the emetogenicity of the chemotherapy administered; included an implicit or explicit definition of complete acute CINV response; described the antiemetic regimen in full; and reported the complete acute CINV response rate as a proportion. The findings of several randomized trials were used to update recommendations for the prevention of acute CINV.
Drug Profile
|
Name |
Nateglinide |
|
Structure |
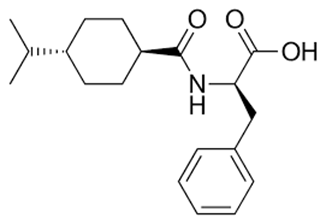
Figure 1.1: Structure of Nateglinide |
|
Molecular Weight: |
317.4 g/mol |
|
Solubility: |
Nateglinide is insoluble in water but soluble in DMSO. |
|
Melting Point: |
137-141 °C |
|
Physicochemical Properties: |
Nateglinide is a white powder. It is freely soluble in methanol, ethanol, and chloroform, soluble in ether, sparingly soluble in acetonitrile and octanol, and practically insoluble in water. Nateglinide capsule-shaped tablets contain 60 mg, or 120 mg, of nateglinide for oral administration. |
|
Uses: |
Nateglinide is used alone or with other medications to control high blood sugar along with a proper diet and exercise program. It is used in people with type 2 diabetes. Controlling high blood sugar helps prevent kidney damage, blindness, nerve problems, loss of limbs, and sexual function problems. Proper control of diabetes may also lessen your risk of a heart attack or stroke. |
MATERIAL AND METHOD
Method Used in the Preparation of Immediate Release Tablets
Direct Compression Method
In this method, tablets are compressed directly from the mixture of the drug and excipients without any preliminary treatment. The mixture to be compressed must have adequate flow properties and cohere under pressure thus making pretreatment as wet granulation unnecessary. Few drugs can be directly compressed into tablets of acceptable quality. A type of disintegrant and its proportion are of prime importance. The other factors to be considered are particle size distribution, contact angle, pore size distribution, tablet hardness and water absorption capacity. All these factors determine the disintegration. The disintegrant addition technology is cost effective and easy to implement at industrial level.
Wet Granulation Method
Wet granulation is a process of using a liquid binder to lightly agglomerate the powder mixture. The amount of liquid has to be properly controlled, as over-wetting will cause the granules to be too hard and under-wetting will cause them to be too soft and friable. Aqueous solutions have the advantage of being safer to deal with than solvent-based systems but may not be suitable for drugs which are degraded by hydrolysis.
Dry Granulation
Dry granulation processes create granules by light compaction of the powder blend under low pressures. The compacts so-formed are broken up gently to produce granules (agglomerates). This process is often used when the product to be granulated is sensitive to moisture and heat. Dry granulation can be conducted on a tablet press using slugging tooling or on a roll press called a roller compactor. Dry granulation equipment offers a wide range of pressures to attain proper densification and granule formation. It is simpler than wet granulation, therefore the cost is reduced. However, this method often produces a higher percentage of fine granules, which can compromise the quality or create yield problems for the tablet. Dry granulation requires drugs or excipients with cohesive properties, and a dry binder' may need to be added to the formulation to facilitate the formation of granules. At last powdered lubricants are added.
Formulation development
Nateglinide Immediate Release tablets were prepared by wet granulation method.
1.1 Table No: Formulation of Nateglinide Tablets
|
Excipients in mg. |
F-1 |
F-2 |
F-3 |
F-4 |
F-5 |
F-6 |
|
Nateglinide |
10 |
10 |
10 |
10 |
10 |
10 |
|
Starch |
54.2 |
59.2 |
54.2 |
52.02 |
59.2 |
64.2 |
|
Lactose |
29.5 |
24.5 |
29 |
24 |
4.5 |
19 |
|
Methyl paraben sodium |
0.18 |
0.18 |
0.18 |
0.18 |
0.18 |
0.18 |
|
Propyl paraben sodium. |
0.12 |
0.12 |
0.12 |
0.12 |
0.12 |
0.12 |
|
Starch for paste |
4 |
4 |
4 |
4 |
4 |
4 |
|
Talc |
1 |
- |
1 |
1 |
- |
1 |
|
Magnesium stearate |
1 |
1 |
1 |
1 |
1 |
1 |
|
Sodium starch glycolate |
- |
- |
0.5 |
1 |
- |
1.5 |
|
Total weight (in mg) |
100 |
100 |
100 |
100 |
100 |
100 |
Preparation of Nateglinide Tablets:
1. Sieving: The active ingredients and the other excipients are passed through sieve no #22. After sieving, they are collected to suitable baskets.
2. Dry Mixing: Above sieved materials are collected and then add into MCG. Dry mixing is achieved by low speed for 15 min. 3.
3. Wet granulation:
i. Preparation of Binder solution:
Take the calculated amount of Starch in a S.S vessel and dissolved it in specified amount of water and then boils at 1000C. Then this solution is added to another solution containing required amount of Methyl Paraben and Propyl Paraben which is heated at 600C. Stir it well.
ii. Mixing:
Add binder solution to the above sieved material which mixing for 5-15 min in MCG in slow speed. Change the mixer speed to fast and mix for 5-15 min till the end point achieved. Then pass through the cad mill to achieve required type of granules. Then scrap the materials from the cad mill.
RESULTS AND DISCUSSION
Organoleptic properties of Nateglinide:
Table no. 1.2: Solubility study of Nateglinide
|
Medium |
Solubility of Nateglinide |
|
Water |
insoluble |
|
DMSO |
5mg/ml. |
Table no.1.3: Melting point of Nateglinide
|
Sr. No. |
Sample |
Melting point |
Reported |
|
1 |
Nateglinide |
137-141 C |
137-141 C |
The standard solutions containing Nateglinide 1 µg/ml was scanned in UV range 200-400 nm using water as blank.
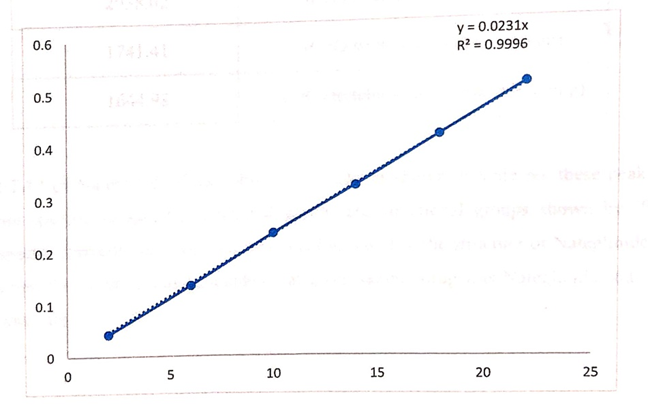
Figure No 1.2: Calibration curve of Nateglinide
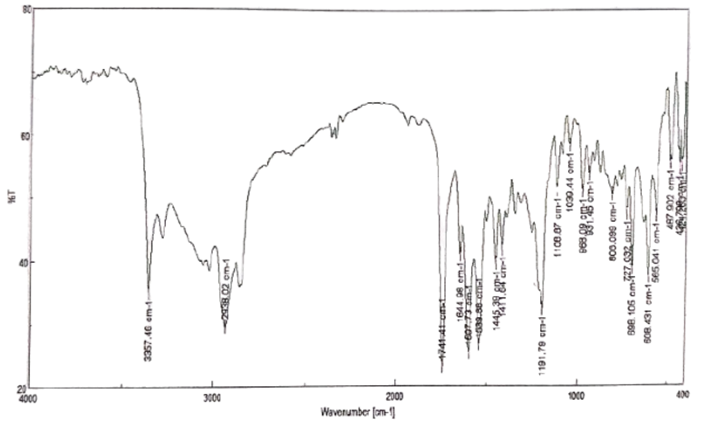
Fig no. 1.3: FTIR Spectra of Nateglinide.

Figure No.1.4: IR spectrum of formulation blend

Figure no.1.5: Spectrum of DSC of Nateglinide
Table no.1.4: Precompression parameters of Nateglinide
|
Batch |
Angle of Repose q |
Bulk density |
Tapped Density. |
Carr's Index 100 x (Dt)-(Db)/ (Dt) |
Hausner Ratio |
|
F1 |
28.44447 |
0.38671138 |
0.405494095 |
5 |
1.05263158 |
|
F2 |
22.692227 |
0.3907511 |
0.403376415 |
2.5 |
1.02564103 |
|
F3 |
25.755567 |
0.38663405 |
0.396923819 |
2.564103 |
1.02631579 |
|
F4 |
24.255557 |
0.38682024 |
0.398974825 |
2.564103 |
1.02631579 |
|
F5 |
25.586667 |
0.38682034 |
0.392723214 |
2.564103 |
1.02631579 |
|
F6 |
25.396667 |
0.38273411 |
0.39695557 |
2.564103 |
1.02631579 |
All batches (F1-F6) were assessed for organoleptic properties like colour, Odor, and taste and found to be acceptable in all aspect.
General appearance: The formulated tablets were assessed for its general appearance and observations were made for shape, colour and texture.
From the results obtained it was found that F1-F6 formulations has hardness, weight variation & friability within IP limit.
Table 1.5: Weight variation test-F1-F6
|
Sr. No. |
Parameter |
F-1 |
F-2 |
F-3 |
F-4 |
F-5 |
F-6 |
|
1 |
Weight variation (%) |
4.59 |
4.50 |
4.58 |
4.52 |
4.50 |
4.59 |
Table 1.6: Tablet parameters (Batch F1-F6) - Thickness.
|
Batch |
Thickness in mm n=3 |
S.D. ± |
|||
|
1 |
2 |
3 |
Mean |
||
|
F1 |
0.2 |
0.21 |
0.22 |
0.21 |
0.01 |
|
F2 |
0.21 |
0.22 |
0.21 |
0.21333333 |
0.0057735 |
|
F3 |
0.22 |
0.21 |
0.22 |
0.21666667 |
0.0057735 |
|
F4 |
0.22 |
0.23 |
0.23 |
0.22666667 |
0.0057735 |
|
F5 |
0.23 |
0.22 |
0.23 |
0.22666667 |
0.0057735 |
|
F6 |
0.23 |
0.23 |
0.22 |
0.22666667 |
0.0057735 |
±S.D. n=3
Table 1.7: Tablet parameters (Batch F1-F6) - Hardness test
|
Batch |
Thickness in mm n=3 |
S.D. ± |
|||
|
1 |
2 |
3 |
Mean |
||
|
F1 |
5.2 |
5.1 |
5.3 |
5.2 |
0.1 |
|
F2 |
5.4 |
5.3 |
5.3 |
5.33333333 |
0.05773503 |
|
F3 |
5.3 |
5.2 |
5.3 |
5.26666667 |
0.05773503 |
|
F4 |
5.3 |
5.2 |
5.3 |
5.26666667 |
0.05773503 |
|
F5 |
5.3 |
5.4 |
5.3 |
5.33333333 |
0.05773503 |
|
F6 |
5.3 |
5.1 |
5.3 |
5.23333333 |
0.11547005 |
±S.D. n=3
Table 1.8. Tablet parameters (Batch F1-F6) - Friability test.
|
Batch |
weight of tablets before test (W1) |
weight of tablets after test (W2) |
Friability % %Friability = [(W1-W2)/W1] × 100 |
|
F1 |
4.49 |
4.48 |
0.222717 |
|
F2 |
4.5 |
4.4 |
2.222222 |
|
F3 |
4.48 |
4.47 |
0.223214 |
|
F4 |
4.51 |
4.5 |
0.221729 |
|
F5 |
4.5 |
4.49 |
0.222222 |
|
F6 |
4.47 |
4.46 |
0.223714 |
Table 1.9. Tablet parameters (Batch F1-F6)-Disintegration time.
|
Batch |
Disintegration Time (min) |
|
F1 |
12.65 |
|
F2 |
13.30 |
|
F3 |
14.45 |
|
F4 |
12.33 |
|
F5 |
14.67 |
|
F6 |
16.12 |
Table 1.10: Dissolution Data
|
Time |
F-1 |
F-2 |
F-3 |
F-4 |
F-5 |
F-6 |
|
0 |
0 |
0 |
0 |
0 |
0 |
0 |
|
10 |
15.22 |
37.22 |
27.08 |
38.18 |
28.44 |
22.28 |
|
15 |
25.17 |
53.21 |
36.69 |
39.90 |
39.54 |
29.14 |
|
30 |
45.55 |
55.34 |
67.44 |
69.53 |
49.22 |
44.56 |
|
45 |
64.33 |
80.14 |
75.85 |
77.26 |
67.52 |
66.35 |
|
60 |
79.81 |
87.16 |
86.17 |
78.59 |
76.67 |
79.89 |
±S.D. n=6
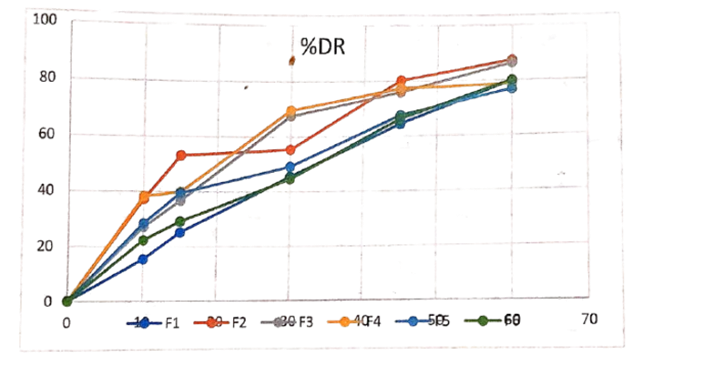
Figure 1.6 -% Cumulative drug release (F1-F6)
Table 1.11: Dissolution Study Data of stability study
|
Time in Min |
Initial |
First month |
|
0 |
0 |
0 |
|
10 |
37.22 |
37.21 |
|
15 |
53.21 |
53.22 |
|
30 |
55.34 |
55.35 |
|
45 |
80.14 |
80.13 |
|
60 |
87.16 |
87.13 |
CONCLUSION
This study successfully developed and assessed immediate release tablets of Nateglinide through the wet granulation technique. Out of the six formulations (F1–F6), formulation F2 delivered the most favourable outcomes regarding disintegration time, hardness, friability, and cumulative drug release. The research demonstrated that the selection and ratio of excipients, particularly disintegrants, play a crucial role in the tablet's performance. Compatibility tests (FTIR and DSC) indicated no interactions between the drug and excipients, ensuring the stability of the formulation. In summary, the optimized immediate release tablet of Nateglinide provides an effective and dependable dosage form for rapid action in managing Type 2 diabetes mellitus.
REFERENCES





Dr. Ganesh Tolsarwad*, Rohini Holkunde, Shripad Ahankari, Sunil Dongre, Gavhane Anjali, Formulation Development and Evaluation of Immediate Releasing Tablet Containing Nateglinide, Int. J. of Pharm. Sci., 2025, Vol 3, Issue 7, 2650-2660. https://doi.org/10.5281/zenodo.16142302
 10.5281/zenodo.16142302
10.5281/zenodo.16142302