
We use cookies to ensure our website works properly and to personalise your experience. Cookies policy
Maharaja Agrasen School of Pharmacy, Maharaja Agrasen University, Baddi, Himachal Pradesh 174103
This study investigates the anticancer potential of three newly designed compound (1a, 1b, and 1c) using molecular docking and in silico ADMET profiling. All compounds demonstrated strong binding affinities toward the target protein, with compound 1b showing the highest docking score (?10.35 kcal/mol), followed by 1a (?9.58 kcal/mol) and 1c (?9.33 kcal/mol). ADMET predictions indicated excellent oral bioavailability, high intestinal absorption, and favorable caco-2 permeability. The compounds exhibited moderate volume of distribution and balanced plasma protein binding, supporting efficient systemic exposure. Despite moderate blood–brain barrier permeability, central nervous system involvement appeared minimal. None of the compounds were predicted to interact with major cytochrome P450 enzymes or OCT2 transporters, reducing the risk of metabolic or renal complications. Toxicity assessments revealed no mutagenic, hepatotoxic, or cardiotoxic effects, though skin sensitization was predicted. Overall, compound 1b emerged as the most promising candidate, warranting further in vitro and in vivo studies to confirm its therapeutic potential.
Cancer is a multifaceted disease characterized by uncontrolled cell proliferation, metastasis, and the ability to evade normal regulatory mechanisms. Among the various subtypes, breast and liver cancers rank among the most prevalent and lethal worldwide. According to GLOBOCAN 2020, breast cancer has become the most commonly diagnosed cancer globally, accounting for over 2.3 million new cases annually, while liver cancer remains the third leading cause of cancer-related mortality due to its aggressive nature and poor prognosis (1).
MCF-7 is a well-established human breast cancer cell line derived from a metastatic site of breast adenocarcinoma. It is widely used in cancer research due to its hormone receptor-positive status, particularly estrogen receptor alpha (ERα), which makes it a suitable model for hormone-responsive breast tumors (2). Similarly, HepG2 is a human hepatocellular carcinoma (HCC) cell line that retains many of the metabolic and functional features of normal hepatocytes. These cell lines provide robust platforms for preclinical evaluation of new anticancer agents.
Both MCF-7 and HepG2 cells share a common molecular feature: aberrant activation of receptor tyrosine kinases and downstream pathways, particularly the PI3K/Akt/mTOR and c-Met/HGF signalling cascades. These pathways play pivotal roles in promoting tumor cell proliferation, angiogenesis, survival, and metastasis, making them attractive targets for therapeutic intervention (3, 4). Natural products and their derivatives have long served as blueprints for anticancer drug discovery. Among these, polycyclic ether frameworks such as 2,5,5,8a-tetramethyloctahydro-2H-chromene offer a unique combination of semi-rigid conformation, lipophilicity, and synthetic accessibility, making them ideal candidates for medicinal chemistry exploration.
Although 2,5,5,8a-tetramethyloctahydro-2H-chromene itself has not been extensively studied in cancer research, its structural resemblance to biologically active chromenes, tetrahydronaphthalenes, and thiazole derivatives—many of which display potent cytotoxic effects—suggests untapped potential. Modifying this scaffold allows the introduction of pharmacophoric groups that can interact with kinase binding pockets, potentially improving target specificity and drug-likeness.
In silico drug discovery has emerged as an essential step in the early stages of lead identification and optimization. Computational approaches such as molecular docking, pharmacophore modeling, QSAR analysis, and ADMET prediction can efficiently prioritize candidate molecules, reduce experimental costs, and streamline the drug development pipeline (5). Molecular docking provides insights into how a ligand binds within the active site of a target protein, estimating binding energy and identifying key interactions such as hydrogen bonding, hydrophobic contacts, and π-π stacking. In the context of PI3Kα and c-Met, docking allows the identification of molecules capable of fitting into the ATP-binding clefts and disrupting kinase activity.
ADMET profiling—which encompasses Absorption, Distribution, Metabolism, Excretion, and Toxicity—helps evaluate the drug-likeness and pharmacokinetic behavior of compounds. Tools such as SwissADME and PreADMET provide in silico predictions on important properties like blood–brain barrier (BBB) permeability, human intestinal absorption (HIA), plasma protein binding (PPB), and mutagenicity. These evaluations are crucial to eliminate molecules with poor bioavailability or high toxicity before advancing to synthesis or biological assays (6).
In this present study, novel derivatives of 2H- chromene were evaluated for their binding potential against PI3Kα and c-Met using molecular docking, assessing their drug-likeness and safety profiles through ADMET prediction and identify lead candidates for potential further development as dual-action anticancer agents for breast and liver cancer therapy. This research utilizes an entirely computational approach, focusing on ligand design, docking validation, and pharmacokinetic modeling to provide a rationale for further experimental exploration.
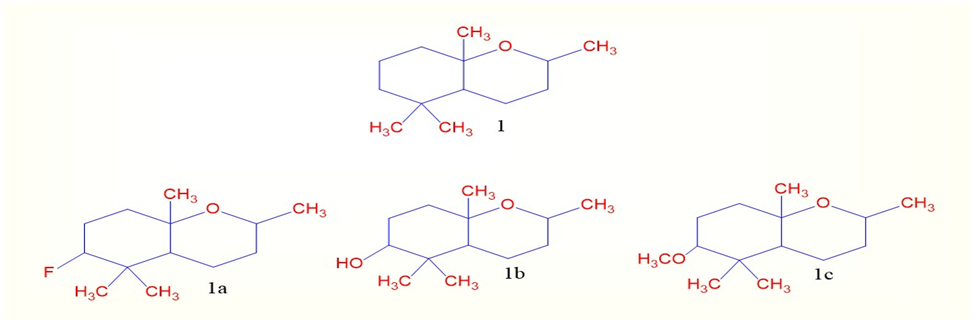
Figure 1 Structures of 2H-chromene derivatives
MATERIALS AND METHODS
In Silico Tools and Computational Setup
All computational analyses were conducted on a Windows 11 pro (64-bit) system with Intel® Core™ i5 processor and 8 GB RAM. Molecular docking was performed using ArgusLab 4.0.1, a free molecular modeling tool used for ligand optimization, receptor grid generation, and flexible docking to predict binding affinities.
For ADMET prediction, the PKCSM online tool (http://biosig.unimelb.edu.au/pkcsm/) was used. It provides predictions for key pharmacokinetic and toxicity properties, including absorption, distribution, metabolism, excretion, and toxicity parameters. Ligand structures were prepared in .mol or .pdb format using ArgusLab or retrieved from databases like PubChem.
Molecular Docking Using ArgusLab
A novel series of Compound 1 (1a, 2b and 3c) was designed as shown in Figure 1 based on structural modifications to the core compound 1 scaffold. Substitutions were introduced at key positions to modulate electronic distribution, steric hindrance, and hydrogen bonding potential—factors known to influence biological activity and pharmacokinetics.
Molecular docking studies were carried out using ArgusLab 4.0.1 to evaluate the binding affinity of three synthesized derivatives with the estrogen receptor alpha (ERα) protein associated with the MCF-7 breast cancer cell line. The crystal structure of the target protein (PDB ID: 3ERT) was retrieved from the RCSB Protein Data Bank (https://www.rcsb.org). The protein was prepared by removing water molecules and any co-crystallized ligands. Hydrogen atoms were added, and the structure was energy-minimized using default settings (7).
The chemical structures of the three derivatives were drawn and optimized using ArgusLab’s geometry optimization tool. Each ligand was saved in .mol format and converted to the appropriate format for docking. Docking was performed using the ArgusDock algorithm with a grid box defined around the active site of the protein. The docking parameters were set to allow full ligand flexibility, while the receptor remained rigid. The binding site was selected based on the co-crystallized ligand's position in the 3ERT structure. Each ligand was docked individually with the prepared protein, and docking scores (binding energies) were recorded. The best binding pose for each compound was selected based on the lowest binding energy and proper orientation within the active site.
Predictive Modeling of Drug-Like Properties and Toxicological Profiles
The pharmacokinetic properties and toxicity profiles of the selected derivatives were predicted using SwissADME (http://www.swissadme.ch/) and PKCSM (http://biosig.unimelb.edu.au/pkcsm/). SwissADME was employed to evaluate key drug-likeness criteria, including Lipinski’s rule of five (8, 9).
For comprehensive ADMET profiling, the compounds were further analyzed using the PKCSM online tool, which predicts absorption, distribution, metabolism, excretion, and toxicity based on graph-based molecular signatures. Parameters such as intestinal absorption, CYP450 enzyme inhibition, total clearance, hepatotoxicity, and AMES toxicity were assessed to evaluate the pharmacological suitability and safety of the compounds (10).
SMILES representations of each compound were used as input for both tools.
RESULTS AND DISCUSSION
Molecular Docking Results
Molecular docking was carried out to assess the binding affinities of 3 derivatised compounds against 3-ERT protein. The binding energies of all compounds was summarized in Table 1.
ADMET Prediction
The development of drugs intended to treat various life-threatening diseases is often both time-intensive and financially demanding. In recent years, the integration of computational tools with multidisciplinary scientific approaches has played a crucial role in streamlining the process of novel drug design. While drug-likeness evaluations do not offer definitive conclusions, they serve as valuable predictive tools during the initial phases of drug discovery. These evaluations rely on established criteria derived from previously approved pharmaceuticals. Among the most widely applied guidelines in this context is Lipinski’s Rule of 5, which helps assess the oral bioavailability and drug-like behavior of candidate molecules. The drug-likeness of all compounds (1,1a,1b and 1c) was assessed based on Lipinski’s Rule of Five, with the results summarized in Table 1. Drug likeness results shows that neither compound exhibits any violations of the established criteria, suggesting their potential suitability for biological use without limitations related to basic physicochemical properties.
Table 1: Drug likeness analysis of compounds 1, 1a, 1b and 1c
|
Lipinski’s 5 creteria |
Accepted range |
Compound 1 |
Compound 1a |
Compound 1b |
Compound 1c |
|
Molecular weight (g/mol) |
≤500 |
196.33 |
214.32 |
212.333 |
226.36 |
|
HBA |
≤10 |
1 |
2 |
2 |
2 |
|
HBD |
≤5 |
0 |
0 |
1 |
0 |
|
LogP |
≤5 |
3.02 |
2.90 |
2.7411 |
3.3952 |
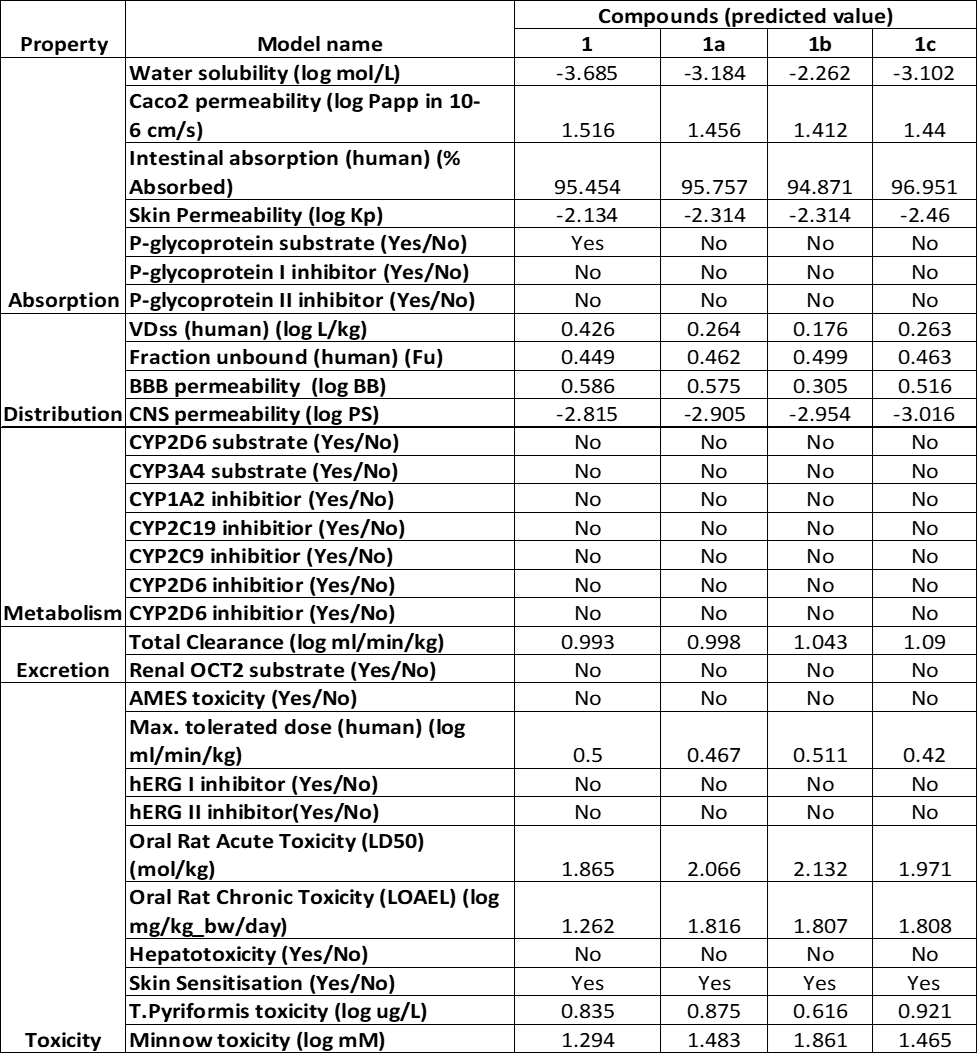
The pharmacokinetic behavior and safety profile of compounds 1, 1a, 1b, and 1c were evaluated in silico by using pkCSM simulations and are shown in Table 2. All four compounds demonstrated excellent oral absorption potential, with human intestinal absorption rates surpassing 94%, indicating strong likelihood of efficient gastrointestinal uptake. Consistently, each compound also exhibited favorable permeability across Caco-2 cell models, supporting their capacity for effective absorption in the human intestine. In terms of distribution, the compounds showed moderate volume of distribution (log VDss values between 0.176 and 0.426), suggesting reasonable tissue penetration. The fraction of unbound drug in plasma ranged from 0.449 to 0.499, which reflects good systemic bioavailability. Despite displaying moderate to high blood–brain barrier (BBB) permeability (log BB > 0.3), their low central nervous system (CNS) penetration scores imply minimal brain exposure—potentially limiting unwanted CNS effects.
Metabolically, none of the candidates were predicted to act as substrates or inhibitors of the key cytochrome P450 enzymes (CYP3A4, CYP2D6, CYP2C9), reducing the likelihood of metabolic interactions or variability in drug clearance. Regarding elimination, total body clearance values were within acceptable pharmacological limits, and none of the compounds were identified as renal OCT2 transporter substrates. Toxicity evaluations revealed no mutagenic activity (via the Ames test), no hepatotoxicity, and no inhibition of hERG channels, suggesting favorable cardiac and liver safety. However, all four compounds were predicted to cause skin sensitization. Acute and chronic toxicity models in rats indicated acceptable safety margins, and environmental toxicity assessments showed moderate toxicity toward T. pyriformis and minnows.
Table 3: Docking Score Interaction between 3 derivative compounds and human estrogen receptor (PDB:3ERT, PDB DOI: (https://doi.org/10.2210/pdb3ERT/pdb)
|
Compounds |
Docking Score/Binding Energy (kcal/mol) |
|
1a |
-9.58 |
|
1b |
-10.35 |
|
1c |
-9.33 |
The molecular docking analysis showed that each compound demonstrated strong binding within the active site, indicating favorable molecular interactions. Notably, compound 1b exhibited the highest binding affinity, with a docking energy of −10.35 kcal/mol, pointing to a highly stable and well-fitted complex. This was followed by compound 1a, which recorded a docking score of −9.58 kcal/mol, and compound 1c, which showed a slightly weaker yet still effective score of −9.33 kcal/mol. The docking result are shown in Table 3. The interaction of ligands with proteins are picturized in Figure 2.
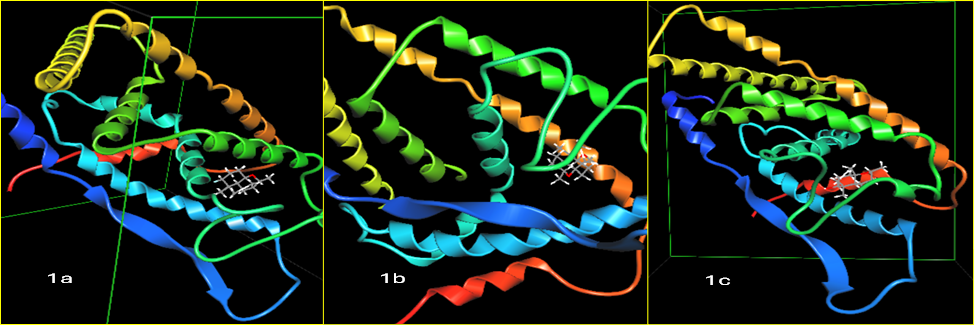
Figure 2 Interaction of 2H -chromene derivatives with 3-ERT
Table 2 ADMET results retrieved from pkCSM tools
CONCLUSION
This study focused on evaluating the anticancer potential of three novel compounds—1a, 1b, and 1c—using molecular docking and ADMET-based computational approaches. Docking analysis revealed strong binding affinities across all three molecules, with compound 1b displaying the most potent interaction (−10.35 kcal/mol), followed by compound 1a (−9.58 kcal/mol) and compound 1c (−9.33 kcal/mol). These results indicate favorable binding within the target’s active site, suggesting these compounds could serve as effective inhibitors in cancer therapy.
ADMET predictions further supported their potential drug-likeness. All compounds showed high intestinal absorption and good Caco-2 cell permeability, indicating suitability for oral administration. The compounds exhibited moderate distribution in body tissues and maintained a balanced fraction of unbound drug in plasma, which supports systemic availability. Although moderate to high blood–brain barrier permeability was observed, their low CNS penetration scores reduce concerns related to central nervous system side effects.
None of the compounds were found to be substrates or inhibitors of major cytochrome P450 enzymes, lowering the likelihood of metabolic interactions. Their excretion profiles were within desirable limits, and none were linked to OCT2 transporter-mediated renal clearance. Toxicological assessments revealed no signs of mutagenicity, hepatotoxicity, or cardiotoxicity (via hERG inhibition), though all three compounds were predicted to cause skin sensitization.
Overall, the combination of strong binding affinity and favorable pharmacokinetic and safety profiles highlights compounds 1a, 1b, and 1c as viable leads for further exploration. Notably, compound 1b emerged as the most promising candidate, meriting additional experimental studies to validate its therapeutic potential in cancer treatment.
REFERENCES

Arvinder Pal Singh, Mona Piplani, Amit Aggarwal, In Silico Design, Molecular Docking, and ADMET Prediction of Novel 2H-Chromene Derivatives as Potential Dual Inhibitors Against MCF-7 Cancer Cell Lines, Int. J. of Pharm. Sci., 2025, Vol 3, Issue 8, 412-418. https://doi.org/10.5281/zenodo.16738011
 10.5281/zenodo.16738011
10.5281/zenodo.16738011