Department of Pharmaceutical chemistry, Prime College of Pharmacy, Prime nagar, Palakkad, Errattayal, Kerala, India
For more than three decades, the generation of therapeutically significant small molecules has been greatly aided by computer-aided drug discovery and design techniques. These techniques can be broadly classified as structure-based or ligand-based techniques. In theory, high-throughput screening and structure-based approaches are similar in that both target and ligand structure information are relevant. Ligand docking, pharmacophore, and ligand design techniques are examples of structure-based methods. Cancers are a large family of diseases which involve abnormal cell growth with the potential to invade or spread to other parts of the body. They form a subset of neoplasms. A neoplasm or tumor is a group of cells that have undergone unregulated growth, and will often form a mass or lump, but may be distributed diffusely. CADD is typically used in drug discovery campaigns for three main reasonsto reduce large compound libraries into manageable sets of predicted active compounds that can be tested experimentally; to direct the optimisation of lead compounds, whether to boost their affinity or optimise drug metabolism and pharmacokinetics (DMPK) properties, including absorption, distribution, metabolism, excretion, and the potential for toxicity (ADMET) and to design novel compounds, Finally, optimisation of the target and therapeutic compounds' advantageous physiological features using computational approaches for toxicity prediction.
Drug discovery is a process of invention that identifies new potential drug candidates. It involves a vast array of clinical specialties, including pharmacology, chemistry and biology. A drug discovery process typically takes 10 to 14 years1. The processes are typically subdivided into preclinical and clinical evaluation, disease target identification, target validation, virtual screening and lead optimization. The investigation of new candidate drugs toxicity, effectiveness, and safety is the core emphasis of drug development. The primary goal of drug development is the creation of scientific evidence that supports a drug safety profile and the dosage that is intended for marketing2.
Different Stages of drug discovery and development include:
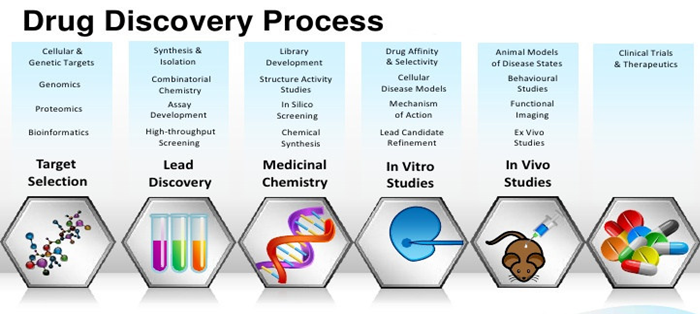
Fig.1:DRUG DISCOVERY PROCESS
1.2.1.Target identification
Target based drug discovery begins with the identification of the function of a potential therapeutic drug target and understanding its role in the disease process3.A target is generally a single molecule, such as a gene or protein, which is involved in a particular disease. A drug target is a key molecule involved in a particular metabolic or signaling pathway that is specific to a disease condition or pathology, or to the infectivity or survival of a microbial pathogen. Some approaches attempt to inhibit the functioning of the pathway in the diseased state by causing a key molecule to stop functioning. Drugs may be designed that bind to the active region and inhibit this key molecule. Another approach may be to enhance the normal pathway by promoting specific molecules in the normal pathways that may have been affected in the diseased state4. The exact target and the specific patient population are identified with the help of bioinformatics, cheminformatics and/or data maiming approaches such as, homology based, ligand based structures based, high throughput screening (HTS), text mining, microarray technologies, pattern matching etc.
1.2.2 Target validation
After a drug target has been identified, a rigorous evaluation needs to occur to demonstrate that modulation of the target will have the desired therapeutic effect. In the drug-discovery process, the major bottleneck is target validation. If this process can be accelerated with computational tools, the target validation step will speed up significantly. The target-validation process includes determining if the modulation of a target’s function will yield a desired clinical outcome, specifically the improvement or elimination or a phenotype. In silico characterization can be carried by using approaches such as genetic-network mapping, protein-pathway mapping, protein–protein interactions, disease-locus mapping, and subcellular localization predictions. Initial selection of a target may be based on the preliminary results found between cellular location and disease/health condition, protein expression, potential binding sites, cross-organism confirmation, or pathways involved in a disease/health condition5.
1.2.3 HIT and lead identification
For many targets in drug discovery, the identification of a small molecule ‘hit’ as a starting point for the hit-to lead process. The identification of small molecule modulators of protein function and the process of transforming these into high-content lead series are key activities in modern drug discovery6. The “hit-to-lead” phase is usually the follow up of high-throughput screening (HTS). Hits can be identified by one or more of several technology-based approaches like high-throughput biochemical and cellular assays, assay of natural products, structure-based design, peptides and peptidomimetics, chemogenomics and virtual HTS, and literature- and patent-based innovations7.To develop efficient drug discovery practices, it is useful to consider the various strategies that have been reported for hit and lead identification; assay development, where the target is converted to an HTS assay system.
1.2.4 Lead optimization
Lead optimization is the complex, no-linear process of refining the chemical structure of a confirmed hit to improve its drug characteristics with the goal of producing drug candidate. Lead structures are optimized for target affinity and selectivity. Docking techniques are currently applied to aid on structure-based absorption, distribution, metabolism and excretion (ADME). Drug candidates discovered using this approach needs to be validated on a disease-specific animal model to provide experimental proof of concept. This radical shift in the drug discovery process from physiology-based approach to target-based approach offers high screening capacity and supports to formulate simple, clear requirements to candidate drugs, which allows implementation of rational drug design8.
1.2.5 pre-clinical testing
Preclinical studies and testing strategies with and without the use of animal testing methods have the purpose of limiting risks whenever a new active substance is to be used as a medicinal product in humans. They should be designed in such a way as to achieve as early, risk-free, unproblematic, and economic a transition as possible from preclinical to clinical trials in medicinal products development9. Scientists carry out in vitro and in vivo tests. In vitro tests are experiments conducted in the lab, usually carried out in test tubes and beakers (“vitro” is “glass” in Latin) and in vivo studies are those in living cell cultures and animal models (“vivo” is “life” in Latin). Preclinical testing involves: pharmacology, toxicology, pre-formulation, formulation, analytical and pharmacokinetics.
1.2.6 Clinical testing
A clinical trial (also clinical research) is a research study in human volunteers to answer specific health questions. Carefully conducted clinical trials are the fastest and safest way to find treatments that work in people and ways to improve health. During the clinical trial, the investigators: recruit patients with the predetermined characteristics, administer the treatment(s), and collect data on the patients' health for a defined time period. The U.S. National Institutes of Health (NIH) organizes trials into five (5) different types: prevention trials, screening trials, diagnostic trials, treatment trials, quality of life trials and compassionate use trials or expanded access9.
1.2.7 NDA and FDA approval
The new drug application (NDA) is the vehicle in the United States through which drug sponsors formally propose that the food and drug administration (FDA) approve a new pharmaceutical for sale and marketing. The goals of the NDA are to provide enough information to permit FDA reviewers. The NDA includes all of the information from the previous years of work, as well as the proposals for manufacturing and labeling of the new medicine. FDA experts review all the information included in the NDA to determine if it demonstrates that the medicine is safe and effective enough to be approved9.
1.3 COMPUTER AIDED DRUG DESIGN
The concept of computer-aided drug discoverywas developed in the 1970s and popularized by Fortune magazine in 198110. Computer-aided drug discovery has been around for decades, although the past few years have seen a tectonic shift towards embracing computational technologies in both academia and pharma. This shift is largely defined by the flood of data on ligand properties and binding to therapeutic targets and their 3D structures, abundant computing capacities and the advent of on-demand virtual libraries of drug-like small molecules in their billions. Taking full advantage of these resources requires fast computational methods for effective ligand screening11.
1.3.1 IN-SILICO DRUG DISCOVERY PROCESS
Drug discovery process is a critical issue in the pharmaceutical industry since it is a very cost-effective and time consuming process to produce new drug potentials and enlarge the scope of diseases incurred. Drug target identification, being the first phase in drug discovery is becoming an overly time consuming process. In many cases, such produces inefficient results due to failure of conventional approaches like in vivo and in vitro to investigate large scale data. Sophisticated in silico approaches has given a tremendous opportunity to pharmaceutical companies to identify new potential drug targets which in turn affect the success and time of performing clinical trials for discovering new drug targets. The main goal of this work is to review in silico methods for drug discovery process with emphasis on identifying drug targets, where there are genes or proteins associated with specific diseases12. In silicois an expression used to mean "performed on computer or via computer simulation 13.In silico drug designing is a form of computer-based modelling whose technologies are applied in drug discovery processes. Unlike the historical method of drug discovery, by trial and-error testing of chemical substances on animals, and matching the apparent effects to treatments, in silico drug design begins with a knowledge of specific chemical responses in the body or target organism and tailoring combinations of these to fit a treatment profile14.
1.3.2 Types of Computer-aided drug design15
Computer Aided Drug Design can be categorized as two types
The 3D structure of the protein, enzyme, RNA or other macromolecule was determined by X-ray crystallography or nuclear magnetic resonance experiments and it was obtained from the Protein Data Bank (RCSB) for structure based drug design as an alternative, homology modelling techniques (MODELLER or an on-line web server SWISS-MODEL)
b)Ligand Based Drug Design (LBDD)
In the absence of the desired 3D structure, ligand-based drug design is designed to examine how a compounds structure and physicochemical properties correlate to its biological activity. The molecules with enhanced biological activity are predicted being used16
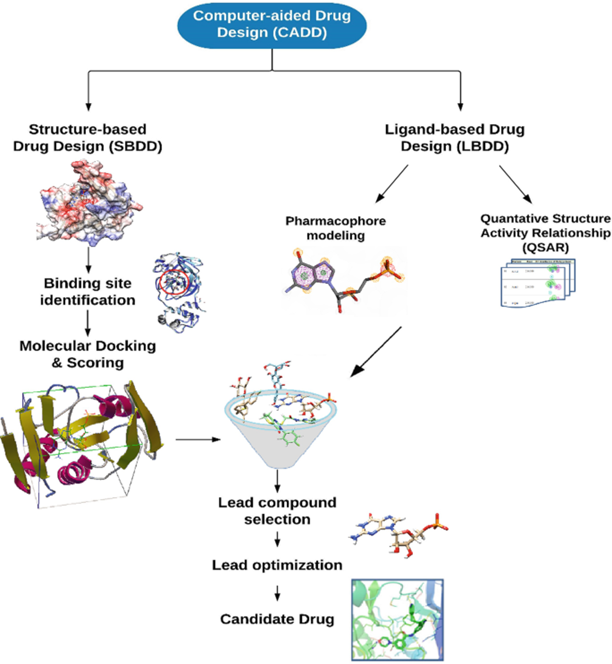
Fig.2: COMPUTER-AIDED DRUG DESIGN
1.3.3 Molecular Docking
Molecular docking is an important component of the drug discovery process and developed in the 1980s, it is used to increasing the number of access to small molecule and protein structures have contributed to the development of improved techniques, carried docking more popular in both industrial and academic field. Molecular docking is the method of how two or more molecular structures (drug and enzyme/ protein or receptor) fit together. Docking is a molecular modeling technique that is used to predict how a protein (enzyme) interacts with small molecules17.
Rigid docking
This approach eliminates any flexibility byv treating the ligand and receptor as rigid entities and exploring only six degrees of translational and rotational freedom. The rigid body docking process is used as the first step in the majority of docking suites.
Flexible docking
A more common technique is tov represent the flexibility of the ligand while assuming that the protein receptor is rigid, thereby only allowing for the ligand's conformational space.
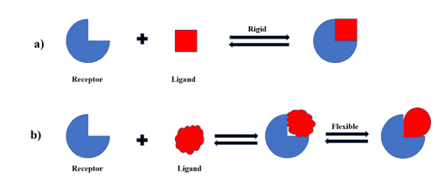
Rigid docking and flexible docking
Active site
Fig.3:DOCKING MODEL
1.4 CANCER
In the last decade, cancer has become one of the most life-threatening diseases, ravaging the world today18,19. It is a major health problem associated with uncontrolled or abnormal cell growth in the body, led to >10 million deaths worldwide in 202020,21.Due to immense side-effects associated with chemotherapy and acquired resistance to available drug regimens as a result of genetic mutation of targets, there is an emerging need to design and develop lead candidates with minimal side effects along with higher potency22-24.Cell growth takes place due to several growth factors in cancerous conditions which may be associated with some abnormality25. The different growth factors such as vascular endothelial growth factor receptor (VEGFR), epidermal growth factor receptor (EGFR), and human epidermal growth factor receptor 2 (HER-2) present on the cell surface transmit signals out of the exterior domain to the interior of the cell26-28. This process is critical as well as essential for multicellular species because cell nuclei require them for optimal cellular activity. The growth factors are made up of polypeptides and their function is to facilitate proliferation and/or differentiation processes in normal cells as well as tumor cells29.EGFR is the first growth factor discovered by Stanley Cohen American biochemist in 1968 and the prototypal member also known as HER (human EGF receptor and c-erbB1) possesses tyrosine kinase activity30. EGFR is composed of a polypeptide chain of 1186 amino acid residues and there are three binding areas present in the EGFR cavity that are extracellular ligand-binding area, the intracellular area with tyrosine kinase (TK) potential, and a hydrophobic region. EGFR is one of the chief successful targets in many cancer therapies, including lung and breast cancers.
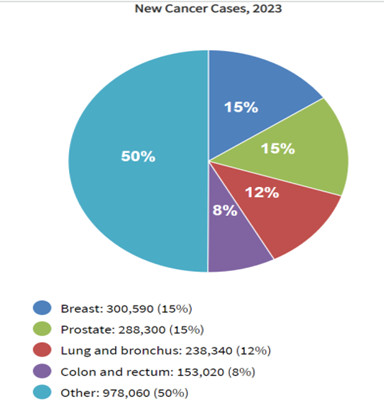
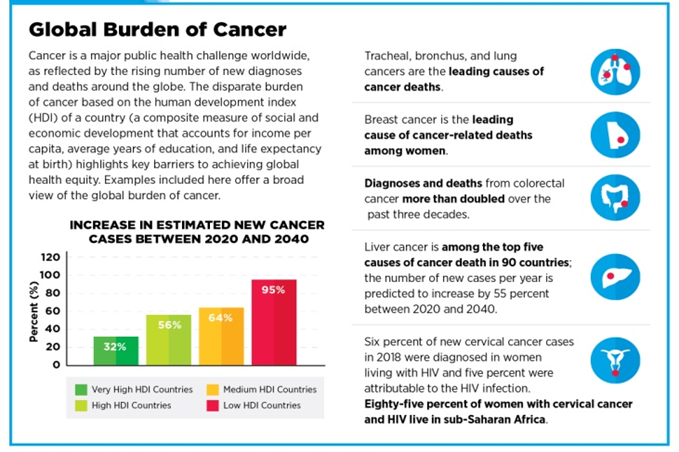
Fig.4: GLOBAL BURDEN OF CANCER
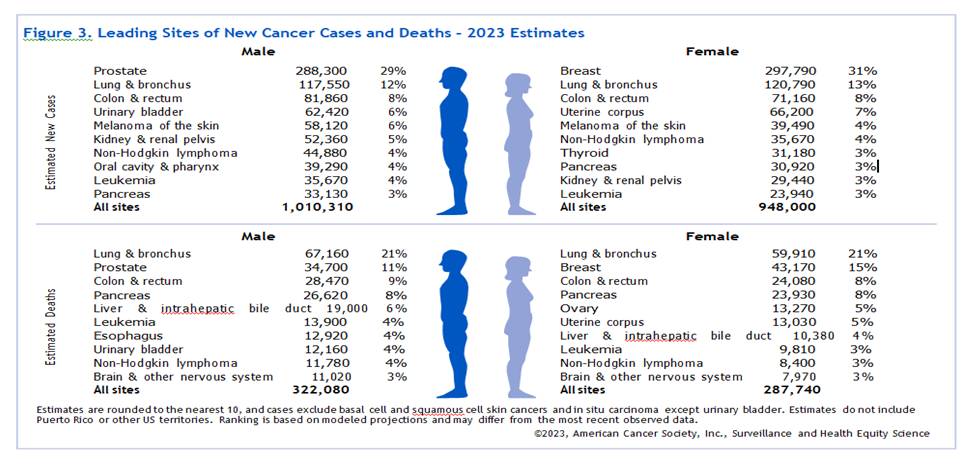
Fig.5: SURVEY REPORT FOR TARGETED CANCER(WHO 2023)
1.4.1 EGFR
Epidermal growth factor receptor (EGFR) is a vital intermediate in cell signaling pathway including cell proliferation, angiogenesis, apoptosis, and metastatic spread and also having four divergent members with similar structural features, such as EGFR (HER1/ErbB1), ErbB2 (HER2/neu), ErbB3 (HER3), and ErbB4 (HER4). EGFR is a transmembrane receptor belonging to a family of four related proteins (Fig. 1).2 Ten different ligands can selectively bind to each receptor. After a ligand binds to a single-chain EGFR, the receptor forms a dimer3 that signals within the cell by activating receptor autophosphorylation through tyrosine kinase activity.3 Autophosphorylation triggers a series of intracellular pathways that may result in cancer-cell proliferation, blocking apoptosis, activating invasion and metastasis, and stimulating tumor-induced neovascularization. Despite this, clinically exploited inhibitors of EGFR (including erlotinib, lapatinib, gefitinib, selumetinib, etc.) are not specific thus provoking unenviable adverse effects. Some of the paramount obstacles to generate and develop new lead molecules of EGF drug resistance, mutation, and also selectivity which inspire medicinal chemists to generate novel chemotypes. The discovery of therapeutic agents that inhibit the precise stage in tumorous cells such as EGFR is one of the chief successfultargets in many cancer therapies, including lung and breast cancers31. The over expression of EGFR has been recognized as the driver mechanism in the occurrence and progression of carcinomas such as lung cancer, breast cancer, pancreatic cancer, etcetera. EGFR receptor was thus established as an important target for the management of solid tumor32.One major family of sensors is comprised of trans membrane receptors with intrinsic protein tyrosine kinase activity (RPTK), the prototypal member of which is the EGF receptor (EGFR; also referred to as HER (human EGF receptor) and c-erbB1) as it was the first receptor described to possess tyrosine kinase activity and the first member of this superfamily to be sequenced33.
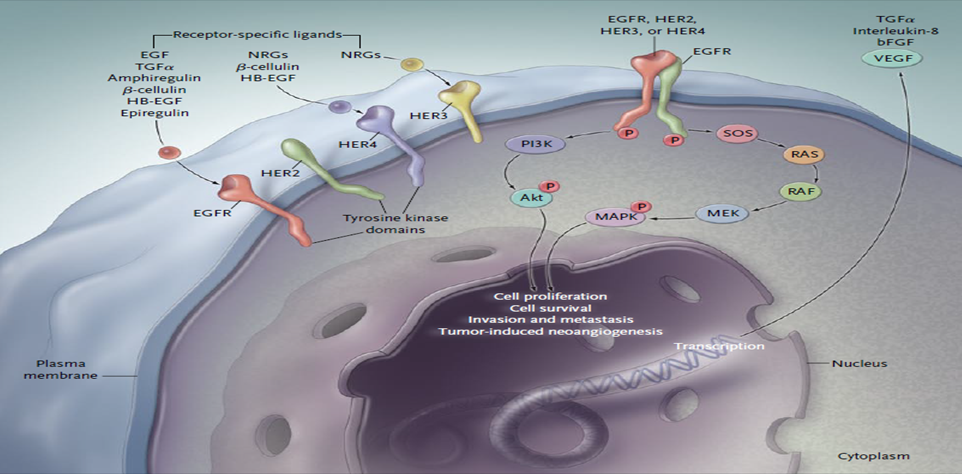
Fig.6: MECHANISM OF EPIDERMAL GROWTH FACTOR RECEPTOR TYROSINE KINASE
1.4.2 Cyclin-dependent kinases- CDK
Cyclin-dependent kinases (CDK) are involved in many crucial processes, such as cell cycle and transcription, as well as communication, metabolism, and apoptosis.Deregulation of any of the stages of the cell cycle or transcription leads to apoptosis but, if uncorrected, can result in a series of diseases, such as cancer, neurodegenerative diseases (Alzheimer’s or Parkinson’s disease), and stroke. CDK-inhibiting drugs have been developed as cancer therapeutics34.The cell cycle is the process by which mammalian cells regulate proliferation and has S, M, G2 and G1 phase. Loss of these cell-cycle control and increased resistance to apoptosis (programmed cell death) represent major hallmarks of cancer. Cyclic dependent kinases (CDKs), a family of serine/threonine, can control the cell cycle progression and transcription. Besides, they are also involved in regulating mRNA processing, the differentiation of nerve cells and glucose homeostasis. Therefore, CDKs are multiple function proteins. Cellular proliferation, driven by CDKs and their cyclin partners, is decontrolled in cancer; therefore, cancer is considered as a proliferative disorder and targeting the cell cycle, therefore, seems to be a good strategy for new targeted anticancer agents. CDKs activity is closely associated with specific cyclin co-factors and at least 12 separate genetic loci are known to code for the CDK35.
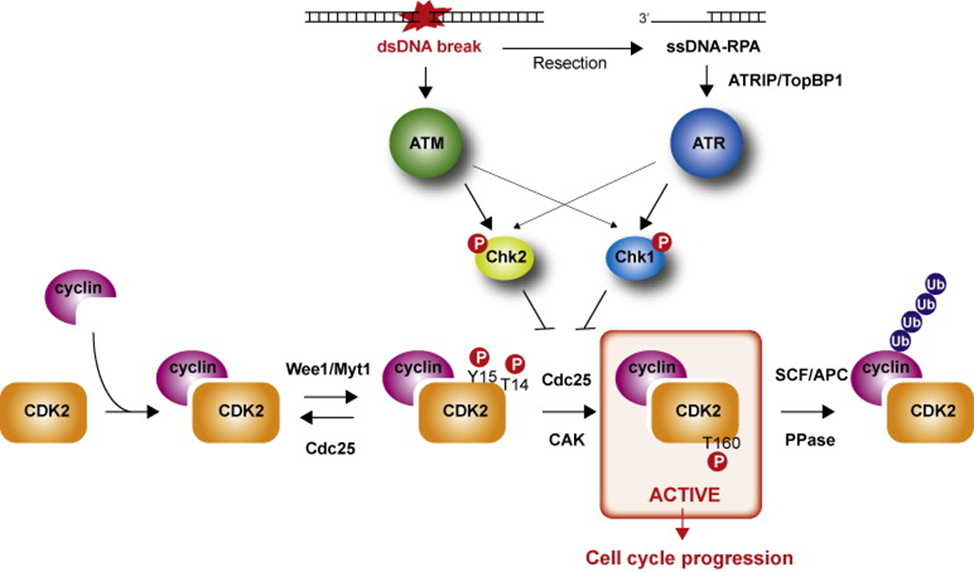
Fig.7: MECHANISM OF CYCLIN DEPENDENT KINASE
1.5 Quinazoline:
Quinazoline is a heterocyclic system having two aromatic six-membered rings. One of them contains two nitrogen atoms named as pyrimidine ring and this ring is fused to the second aromatic benzene ring36 Therefore, quinazoline is a phenyl pyrimidine compound37.
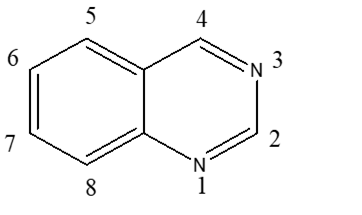
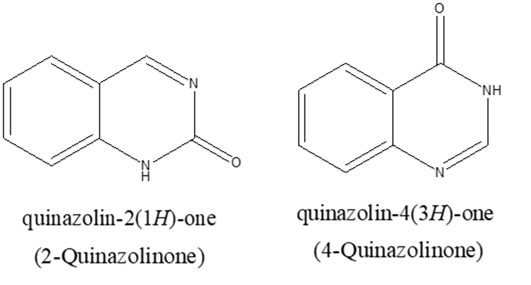
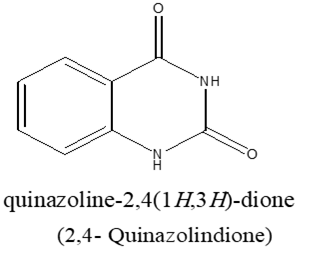
The quinazoline system can be divided into three members, the 2-quinazolinone containing a carbonyl group at the C-2,38. the 4-quinazolinone containing a carbonyl group at the C-439 and the 2,4-quinazolinedione containing two carbonyl groups at the C-2 and the C-440.
Quinazolines have a wide range of pharmacological activities41-43. They are used as anticancer, antiviral, antibacterial, antitubercular, analgesic, antihypertensive, anti-inflammatory, antidiabetic, sedative-hypnotic, antihistaminic, anticonvulsant, and many other uses42-46.
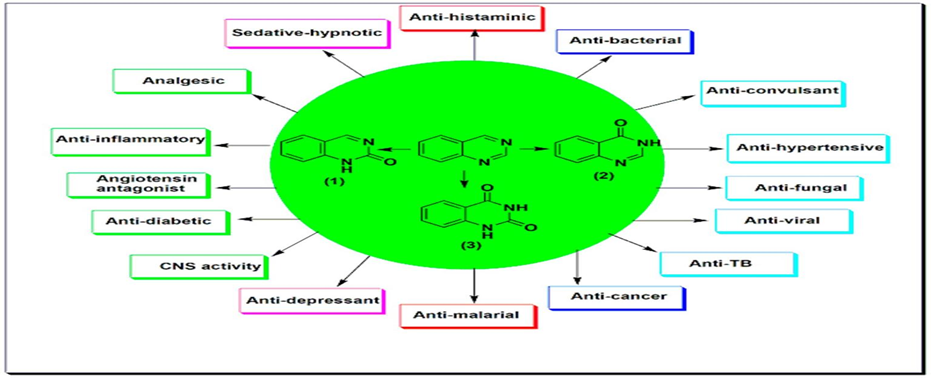
Fig.8: QUINAZOLINE AS A THERAPEUTIC AGENT
Quinazoline molecules form a favoured group of multi acting therapeutic agents in the pharmaceutical and biological fields. The easy preparation and the diverse spectrum of pharmacological activities made this scaffold very important among the different therapeutic agents43 Many of the existing chemotherapeutic agents are highly toxic with a low level of selectivity. Additionally, they lead to development of therapeutic resistance. Hence, the development of targeted chemotherapeutic agents with low side effects and high selectivity is required for cancer treatment. Quinazoline is a vital scaffold well-known to be linked with several biological activities. The anticancer activity is one of the prominent biological activities of this scaffold. Several established anticancer quinazolines work by different mechanisms on the various molecular targets46.
CONCLUSION
This chapter deals with general introduction about drug discovery, phases of drug discovery, CADD, molecular docking and importance of Quinazoline derivatives. Evaluation of the safety, effectiveness, and toxicity of new drug candidates is the main emphasis of the drug discovery process. The development of computational methods , including drug likeness properties, high throughput virtual screening, Insilco ADME characteristics, and molecular docking, has revolutionized the method of drug discovery. Computer-aided drug design is a crucial tool for the rapid quantitative measurement of the properties of potential drug molecules, minimising the need for synthetic and biological testing by choosing to focus only on the most promising molecules, and speeding up the drug design process by considering the structure-activity relationship. The can help in identifying the drug likeness properties and suitable for biological evaluation. Computational techniques used to identify the drug targets, possible active site and binding affinities through bioinformatic tools. Cancer is a primary cause of mortality in worldwide and accounted for 7.4 million deaths (around 13% of all deaths) in 2004. More than 70% of all cancer deaths occurred in low- and middle-income countries. Deaths from cancer worldwide are projected to continue rising, with an estimated 11.5 million deaths in 2030.Globally, the three most common cancers considered are Lung (1,824,701; 13%), Breast (1,676,633; 11.9%) and Colo-rectal cancers (1,360,602; 9.7%) Based on this the main aim and objective of the work was to synthesis, characterize and evaluate the newly selected lead molecule through computational studies against cancer.
REFERENCES


Kavitha K, Niranjana C M, Archamol. V. U, Numra. A. P, Fathima Liyana. K. K, Fousana Sherin, Fahid. A. K, Ali Ahammed H, Gowrishankar N L, Modern Drug Discovery Process of Synthetic Drug in Cancer: A Review, Int. J. of Pharm. Sci., 2026, Vol 4, Issue 3, 3139-3150. https://doi.org/10.5281/zenodo.19217623
 10.5281/zenodo.19217623
10.5281/zenodo.19217623
