
We use cookies to ensure our website works properly and to personalise your experience. Cookies policy
Department of Pharmaceutical Chemistry, Oriental College of Pharmacy, Navi-Mumbai, Maharashtra, India.
Aim: To design a potent inhibitor from natural product source against Mycobacterium tuberculosis using computational studies. Background: Tuberculosis (TB) is one of the world's most deadly infectious disease caused by a single pathogen, ranks second to coronavirus. This highly contagious bacterial illness primarily affects the lungs, it poses significant challenges in drug development, including the emergence of drug tolerance and resistance, potential drug interactions, and adverse effects from existing treatments. In modern pharmaceutical research and development, in-silico methods have become indispensable tools for identifying potential drug candidates. Objective: As conventional anti-tuberculosis drugs face increasing resistance, the demand for novel treatments grows, prompting exploration into alternative avenues such as natural compounds. This study focuses on evaluating the efficacy of natural compounds in combating tuberculosis, leveraging the rich history of herbal medicine in managing pulmonary tuberculosis. Methods: Our findings through molecular docking studies indicate that 3-cinnamoyltribuloside, a natural compound demonstrates promising potential in inhibiting tuberculosis. Through molecular docking simulations, it exhibited the highest docking scores against two first-line anti-tuberculosis drugs, isoniazid and pyrazinamide, as well as the recently approved US-FDA drugs, bedaquiline, delamanid and pretomanid. Notably, its docking score against the isoniazid target, InhA surpassed all others. Results: These results suggest that 3-cinnamoyltribuloside may effectively target key proteins involved in tuberculosis infection, including InhA, pncA, a ATP synthase and DprE1. Conclusion: These in-silico predictions serve as valuable starting points for drug discovery, rigorous experimental validation in biological systems remains crucial to determining efficacy and therapeutic potential.
Tuberculosis (TB) commonly known as “White plague” is a highly contagious life threating infectious disease impacting mostly the lungs and causing the immune system to be compromised and vulnerable to other diseases [1]. Mycobacterium tuberculosis is the causative agent of TB [2]. Mycobacterium tuberculosis is also called as Koch’s Bacillus, since the bacillus isolation and growth was pioneered by Robert Koch on 24th March, 1882 [3, 4]. TB is an airborne disease. When a TB infected person sneezes, coughs, speaks, laughs or even sings the TB germs are dispersed in the surrounding air thus when a healthy person inhales it, the germs can enter into his/her lungs [5]. TB can be prevented and one can even restore back to health but still after Covid-19, TB was second largest cause of demise due to single infectious organism [6]. The capacity of the body to effectively limit or clear the infectious inoculum is influenced by one's immunity, genetic factors, and whether it’s a primary or secondary exposure to organism [7]. Even after numerous medication and various first line therapy drugs combination and Bacillus Calmette-Guérin (BCG) vaccination, still it remains one of the deadliest single pathogen infectious disease present over the globe. Eight countries including Bangladesh (3%), China (8%), India (26%), Indonesia (9%), Nigeria (4%), Pakistan (6%), Philippines (6%) and South Africa (3%) are home to about half of all TB patients [8]. M. tuberculosis is a non-spore-forming, non-motile, obligate-aerobic, facultative, catalase-negative, intracellular bacterium which proliferate slowly for every 18-24 hours in vitro [7]. The cell wall of Mycobacterium tuberculosis has complex and distinctive structure made up of thick peptidoglycan and the outer membrane composed of different lipopolysaccharides and fatty acids with imbedded glycolipids and wax esters. Because of the lipid enrich cell wall of Mycobacterium tuberculosis it creates a low-permeability blockade which guards it against the antibiotics. Thus, being one of the multiple challenges during drug development [9]. It has long chain of β-hydroxylated fatty acids called Mycolic acid, which is one of major targets of anti-TB drugs [10]. The WHO estimates that a person has a 10% lifetime risk of developing tuberculosis. Individuals with compromised immune systems due to HIV, diabetes, malnourishment, or tobacco use are at a higher risk of contracting tuberculosis. TB is curable if detected, diagnose and treated in the early phases [11]. It was estimated that 2.8 million cases of tuberculosis (28.2 lakh) occurred in India in 2022, with a case fatality rate of 13%. As per officials, there were 3,42,000 TB deaths in India (3,31,000 TB deaths among HIV-negative people and 11,000 TB deaths among HIV-positive people). WHO Global TB report 2023 shows that India had highest number of TB cases in the world in 2022, representing 27 percent of the world TB burden [12]. Person with active TB experience symptoms like: Bad cough > three weeks, chest pain, blood cough or sputum, weakness, fatigue, loss of weight, loss of appetite, chills, fever, night sweats [13, 14]. For active TB treatment, following are the first line anti-TB drugs: Isoniazid, Ethambutol, Rifampicin, Ethionamide, Pyrazinamide and Streptomycin. Second line anti-TB drugs include: Injectable aminoglycosides like Amikacin, Kanamycin and Streptomycin. Injectable polypeptides like Capreomycin and Vancomycin. Fluoroquinolones (oral and injectable) like Levofloxacin, Moxifloxacin, Ofloxacin and Gatifloxacin. Others include, Para-amino salicylic acid, Cycloserine, Terizidone, Ethionamide and Prothionamide etc. Third line anti-TB drugs have changing but unproven efficacy against TB. They are used as a last resort in the treatment of total drug-resistant TB and include, Clofazimine, Linezolid, Amoxicillin/clavulanic acid, Imipenem/cilastatin and Clarithromycin [15]. Challenges in the development of anti-TB drugs include the potential for drug tolerance leading to resistance, drug-drug interaction, imperative to shorten treatment duration for drug-sensitive tuberculosis patients, insufficient funding hindering global medical needs, the complexity of the disease, particularly in managing multi-drug regimens, the side effects of the existing anti-TB drugs such as hepatotoxicity of isoniazid, QT interval prolongation of bedaquiline, loss of appetite, nausea, flushing in early regimen of pyrazinamide and other anti-TB drugs leads to the reason for research in the field of anti-TB drugs. Crucially, the on-going resistance to the conventional anti-TB drugs meets the need for development of new anti-tuberculosis drugs [16, 17]. The importance of natural components in the realm of antibacterial therapy is undeniable. Traditionally, natural substances have been crucial for fight against TB. The use of herbs and natural products in traditional medical practice dates back centuries and offers distinct benefits in managing pulmonary TB. Whether administered individually or together with conventional antibiotics demonstrates the antimicrobial effects through a multifaceted approach, targets multiple compounds and pathways. Moreover, they boast significant efficacy, with minimum adverse effects, affordability, and a lower propensity for drug resistance. Extensive clinical and pharmacological investigations have revealed that natural products not only possess notable inhibitory effects on M. tuberculosis, but also induce only mild modulation of the host immune response. Numerous studies have documented the impressive growth inhibitory effects of natural products and their derivatives against M. tuberculosis. Several of these compounds identified as promising candidates for the development of new antitubercular agents, serving as prototypes in ongoing research efforts [18]. Diospyrin, an active compound found in Salvadora persica L, commonly known as the South African toothbrush tree, is renowned for its anti-tuberculosis properties [19]. Similarly, cucurbitacin and ursolic acid components found in Citrullus colocynthis is also known to possess anti-tuberculosis activities [20]. Our control compound, 3-Cinnamoyltribuloside is a chemical compound known as a steroidal saponin has a complex molecular structure that includes a steroid core attached to a sugar moiety and a cinnamoyl group, derived from the Tribulus plant, particularly Tribulus terrestris. It is part of a class of naturally occurring compounds known for their diverse biological activities such as antioxidant, anti-inflammatory, antituberculosis and has potential effects on sexual health, energy levels, and cardiovascular health, also includes potential medicinal and therapeutic benefits [21,22]. Between 1981 and 2010, antibacterial agents derived from natural sources constituted approximately 75% of all discovered drugs. The ongoing investigation into nature's potential as a reservoir for novel antimicrobial agents is broadening, presenting a significant opportunity to uncover diverse chemical scaffolds for drug discovery [23]. Plants serve as a crucial reservoir of secondary metabolites, holding immense therapeutic promise. They continue to be integral to traditional medicinal practices in countries like China and in regions undergoing economic development. Frequently, the knowledge surrounding medicinal plants is transmitted orally across generations without formal documentation or scientific scrutiny. Nonetheless, these plants remain a valuable resource ripe for exploration, potentially yielding "hit" compounds with substantial biological activity—promising leads in drug discovery efforts [24].
2. EXPERIMENTAL
Molecular docking is a computational technique utilized in structural biology and drug discovery predicts the binding mode and strength of interactions between two molecular structures, typically a protein and a ligand molecule (such as a drug or small molecule). It helps to simulate and analyse potential binding configurations and strengths, aiding drug discovery and understanding protein-protein interactions [25, 26]. Process of molecular docking entails several key steps. Initially, protein and ligand structures are prepared for docking. Protein structures are often acquired from experimental techniques like X-ray crystallography or NMR spectroscopy, while ligand structures may be sourced from databases or computationally generated. A three-dimensional grid encompassing the protein's binding site is constructed to facilitate the efficient exploration of potential binding orientations for the ligand within the binding site. A scoring function is deployed to assess the suitability of different ligand conformations at the binding site, factors such as van der Waals interactions, electrostatic interactions, hydrogen bonding, and desolvation energy. Diverse algorithms are employed to navigate the ligand's conformational space within the binding site and predict its optimal binding pose. These algorithms encompass stochastic methods like genetic algorithms and Monte Carlo simulations, as well as deterministic methods like geometric hashing and incremental construction. Following docking, the generated ligand poses undergo scoring using the established function. Poses exhibiting the highest scores are ranked as the most probable binding modes. Predicted binding modes are validated using experimental data, such as mutagenesis studies or biochemical assays. Analysis of the docking results furnishes insights into the binding interactions between the protein and ligand, guiding subsequent drug design endeavours [27, 28] The steps involved in the docking process are as follows: Ligand Preparation (.pdb): To begin the process of importing a ligand into Discovery Studio, firstly we located the canonical SMILES representation of the desired ligand. This can typically be found in databases like Wikipedia or PubMed. Once copied the SMILES notation, opened Discovery Studio and navigated to the "small molecule" panel. From there, selected "new" to initiate the process of adding a new molecule. Next, selected "insert from" and opted for the SMILES option. The copied SMILES notation were pasted into the provided field. After ensuring the correct representation, saved the file by giving it an appropriate name followed by the ".pdb" extension, indicating it's a Protein Data Bank file. This file format is commonly used for storing biomolecular structures. Following these steps will effectively imported the ligand into Discovery Studio for further analysis as needed. Protein Preparation (PDBQT): To prepare the protein for molecular docking, downloaded the protein files in the Protein Data Bank (.pdb) extension format. Once downloaded, opened the desired protein file in the Swiss PDB Viewer application. Selected all components of the protein and initiated energy minimization to optimize its conformation. After energy minimization, saved the current layer of the protein as a new file, replacing the existing .pdb file with an appropriate name for clarity. Next, opened AutoDockTools 1.5.7, specifically version ADT 4.0, and ensured the required startup directory under preferences in the file menu. Within AutoDockTools, deleted any unwanted chemical moieties that are not part of the amino acid chain. Checked for and repaired any missing atoms in the structure, if present. Polar hydrogens added to the protein molecule to ensure accurate representation of its chemical properties. Kollman Charges are then used to allocate partial charges that are applied to the protein's atoms. Following this, assigned AD4 atom types to each atom in the protein structure. Finally, saved the prepared protein file as .pdbqt file, which is the required format for molecular docking simulations. These steps ensured the protein is properly configured and optimized for subsequent docking studies using AutoDockTools. Preparation of Ligand (PDBQT): To integrate the ligand into the docking setup, firstly, ligand file was opened within AutoDockTools by accessing the ligand menu. Once the ligand is loaded, navigated to the ligand options in the ribbon interface. Within this menu, selected the "detect root" option to identify the appropriate root atom for the ligand. After that, the "choose root" option to enter the root atom was selected. This is important because the root atom is used to determine the ligand's orientation during the docking simulation. For the purpose of the ensuing docking analysis, these procedures guarantee precise ligand placement and alignment within the protein binding region. Preparation of Grid (GPF): To configure the docking grid parameters, navigated to the ribbon interface and clicked on the "grid" option. Then, "macromolecules" was selected and specified "protein" to indicate the protein as the target for docking. Next, clicked on "set map types" and the ligand to define the properties for grid generation based on the ligand's characteristics was selected. Subsequently, "grid box" was selected to initiate the creation of the docking grid box. Customized the parameters as needed, such as dimensions and center coordinates, to encompass the binding site of interest. After generating the grid box, ensured to save it for future reference and reproducibility. Finally, saved the overall setup and configuration as an AutoDock Grid Parameter File (.gpf) by selecting "save output" and giving it an appropriate name, typically based on the protein's name, for easy identification and management. This file encapsulates all the necessary information for the docking simulation, including the grid parameters and target protein specifications, facilitating the seamless execution of molecular docking studies. Preparation of Dock files (DPF): To commence the docking simulation, proceed to the ribbon interface and click on the "docking" option. Within this menu, selected "protein" and specified it as rigid, ensuring that its conformation remains unchanged during the docking process. Next, the ligand was selected by clicking on the ligand option and accepting its selection. After defining the macromolecule and ligand, adjusted the search parameters by clicking on "search parameters" and selecting the genetic algorithm, which is a common method for exploring conformational space during docking studies. This algorithm helps optimize ligand binding configurations within the binding site of the protein. Finally, customized the output settings by clicking on "output" and selected Lamarckian Genetic Algorithm (GA), a variant of the genetic algorithm that incorporates adaptive features for enhanced efficiency. Saved the docking parameter file (dpf) with an appropriate name, typically reflecting the protein being studied, to encapsulate all the necessary settings and configurations for the docking simulation. This file serves as a blueprint for the docking process, guiding the software in generating and evaluating ligand-protein interactions. Docking simulation: To execute the docking simulation, navigated to the ribbon interface and clicked on "Run." This initiates the process of running Autogrid, which generates the grid maps necessary for the docking calculations. Within the designated grid box, Autogrid computes the interaction energies between the protein and the ligand. Following the completion of Autogrid, proceeded to run AutoDock, which performs the actual docking simulation. AutoDock explores various orientations and conformations of the ligand within the binding site of the protein, evaluating and scoring each pose based on predicted binding affinity. By completing these steps, the docking simulation was executed, and the software generates results detailing the energetically favorable binding modes of the ligand within the protein's binding site. These results provide valuable insights into ligand-protein interactions, aiding in the design and optimization of potential therapeutic compounds. Analysis of the result: To analyze the results of the docking simulation, navigated to the "Analyze" option in the ribbon interface. From there, opened the .dlg (AutoDock Docking Log) file generated by the docking simulation. This file contains detailed information about the docking results, including the various binding poses and associated energies. Once the .dlg file is opened, clicked on "macromolecule" and selected the protein of interest to visualize its structure within the docking results. Next, navigated to "conformation" to assess the different binding energies of the ligand-protein complexes generated during the simulation. These energies provide insights into the strength of ligand binding and can help identify promising candidate compounds for further study or optimization. By following these steps, we effectively analyzed the docking results and evaluated the potential binding interactions between the Isoniazid and InhA; Pyrazinamide and pncA; Bedaquline and ATP Synthase; Pretomanid and DprE1; Delamanid and DprE1; and control drug 3-cinnamoyltribuloside with all these targets (InhA, pncA, ATP Synthase and DprE1) aiding in drug discovery and development efforts.
3. METHODS
In this study, several software tools were employed to facilitate various computational and visualization tasks related to protein-ligand interactions. Swiss PDB Viewer (SPDBV): It is a software tool used for visualizing and analysing protein structures. Established by the Swiss Institute of Bioinformatics (SIB), SPDBV offers a several features including: Viewing protein structures in various representations (e.g., wireframe, cartoon, space-filling), Analysing protein structures through tools such as measuring distances, angles, dihedral angles, Comparison of Superimposing structures, Editing structures by adding or modifying atoms and residues, Generating molecular surfaces, Sequence alignment performance, Energy minimization and molecular dynamics simulation capabilities. Used to manipulate, analyse, and visualize three-dimensional protein structures obtained from the Protein Data Bank (PDB) [59, 60]. Discovery Studio 2024 client: It is a comprehensive software suite developed by BIOVIA (formerly Accelrys) for computational chemistry, molecular modelling, and simulation tasks in the field of drug discovery and materials science. It was used for molecular visualization, protein modelling, ligand docking, virtual screening, pharmacophore modelling, QSAR (Quantitative Structure-Activity Relationship) analysis, etc. It integrates various algorithms and methodologies for molecular modelling and simulation, allows to explore complex biological systems and design novel compounds with desired properties [61]. ChemDraw: It's a popular software suite used by chemists asnd researchers for drawing chemical structures and reactions. It's developed by PerkinElmer and mostly utilized for creating publication-quality diagrams of molecules and reactions. ChemDraw used to represent chemical structures accurately and efficiently, aiding in communication, analysis, and documentation of chemical information. It provides a wide range of tools and features tailored specifically including templates, atom and bond tools, spectroscopy tools, and integration with databases for retrieving chemical information. ChemDraw facilitated the production of high-quality visuals essential for effectively communicating chemical information [62]. Auto Dock: It is a software application mostly utilized in the realm of computational chemistry and structural biology. It serves as a valuable tool for molecular modelling, predicts the binding modes of small molecules, such as drugs or ligands, to proteins or nucleic acids. Through molecular docking simulations, AutoDock aids in understanding how these molecules interact and bind within specific binding sites on target biomolecules. Using algorithms grounded in molecular mechanics and empirical scoring functions, AutoDock explored various conformations and orientations of the ligand within the binding site to predict energetically favourable binding poses. The software enabled the preparation of receptor and ligand files, definition of the grid box around the binding site, execution of docking simulations, and analysis of binding poses. [63]. MGL Tools: The MGL Tools, an acronym for Molecular Graphics Laboratory Tools, represents a pivotal software suite utilized extensively in molecular modelling and computational chemistry. It aided in the visualization, analysis, and manipulation of molecular structures and simulations. MGL Tools provides a diverse array of functionalities, including molecular visualization, molecular docking, protein structure analysis, and molecular dynamics simulations. It enabled to explore and comprehend complex molecular interactions. MGL Tools essential for preparing input files and analyzing results from AutoDock simulations. ADT allowed for the addition of hydrogens, assignment of charges, preparation of PDBQT files, and definition of grid and docking parameters. Collectively, these software tools enabled a comprehensive analysis of protein-ligand interactions, ensuring robust and reproducible computational experiments that contribute to drug discovery and structural biology research.
4. RESULTS
A novel plant-based secondary metabolite named 3-cinnamoyltribuloside has been identified as possessing anti-tuberculosis activity [40]. Molecular docking studies were conducted to compare its binding efficacy against five known anti-TB drugs: Isoniazid, Pyrazinamide, Bedaquiline, Pretomanid and Delamanid. These drugs, along with their respective targeted proteins were used as reference drugs. Blind docking was performed on both the control group and 3-cinnamoyltribuloside to explore potential binding sites on the entire protein. The PDB IDs of the drugs are as follows: Isoniazid (2nv6), Pyrazinamide (3pl1), Bedaquiline (5yio), Pretomanid (6g83) and Delamanid (6g83). Binding Affinity: The binding affinity (in kcal/mol) of 3-cinnamoyl tribuloside to the target protein was recorded. The docking simulation produced several binding poses for 3-cinnamoyl tribuloside within the active site of the target protein. Lower binding energy indicates a more stable ligand-protein complex. Initially, docking was carried out on one of the standard anti-tuberculosis drugs Isoniazid and natural product 3 cinnamoyltribuloside, revealing that the latter exhibited superior docking scores compared to Isoniazid. Consequently, further docking was conducted with another standard anti-tuberculosis drug Pyrazinamide, where 3-cinnamoyltribuloside demonstrated better scores. To further assess the potential of the natural component 3-cinnamoyltribuloside, docking studies were extended to three recently approved drugs by the United States Food and Drug Administration (US-FDA): Bedaquiline (2012), Delamanid(2014) and Pretomanid (2019).
5. DISCUSSION
Interaction Analysis: The Figure 1 represents the 3D diagram of reference drugs Isoniazid, Pyrazinamide, Bedaquiline, Pretomanid and Delamanid with their respective target proteins InhA, pncA, ATP Synthase, DprE1 and DprE1 also the control drug, 3-cinnamoyltribuloside.

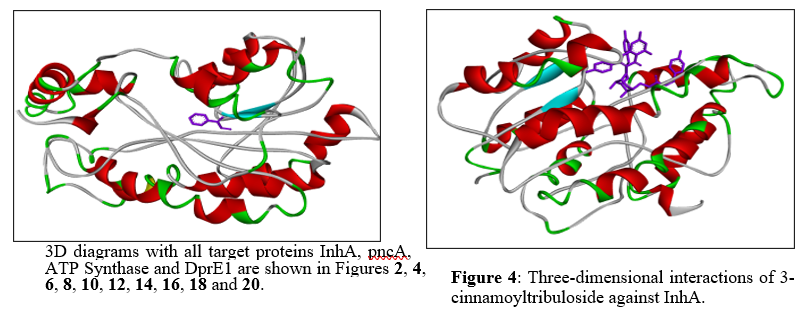
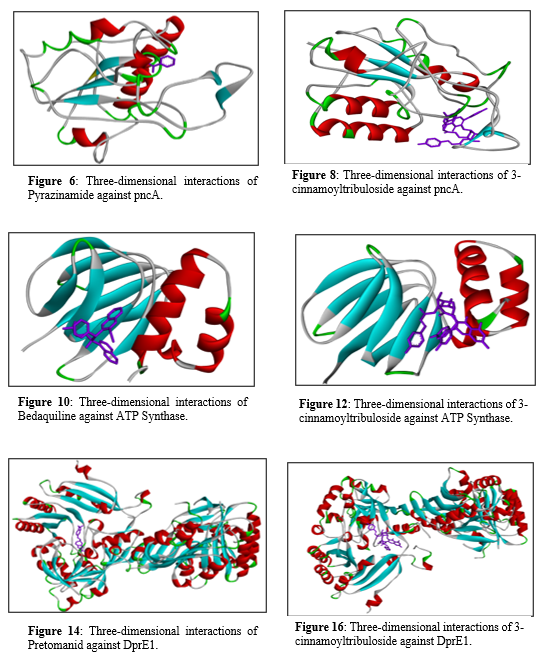
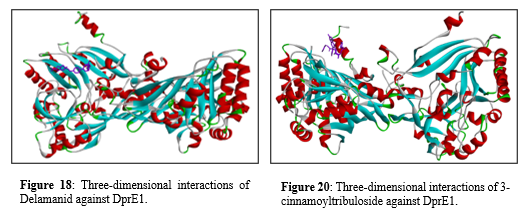
Figure 3 shows two-dimensional interaction of Isoniazid against InhA, Van der Waals interactions with residues such as ILE A:47, ILE A:15, and ILE A:95 interacting with the ligand. GLY A:14 forms a hydrogen bond with the nitrogen atom of isoniazid, contributing to the ligand’s stabilization in the active site. GLY A:40 and THR A:39 form hydrogen bonds with the carbonyl oxygen of isoniazid, enhancing the binding specificity. LEU A:63 forms a hydrogen bond with another nitrogen atom of isoniazid, further stabilizing the complex. ILE A:16 interacts with a carbon atom on isoniazid, providing additional stabilization to the ligand within the binding site. Pi-Alkyl interactions with PHE A:41, ILE A:16 and the aromatic ring of isoniazid.

Figure 4: Three-dimensional interactions of 3-cinnamoyltribuloside against InhA.
Figure 5 shows two-dimensional interaction of 3-cinnamoyltribuloside against InhA where non-covalent Van der Waals interactions (green circles) with residues like MET A:98, MET A:161, LEU A:207 were seen. several hydrogen bonds (green dashed lines) were formed between the ligand and amino acids like PHE A:97, GLN A:100, and ALA A:94, which help in stabilizing the ligand in the binding pocket. Carbon Hydrogen bonds (grey dashed lines) involve residues like ALA A:198 and GLY A:14 interacting with the ligand. Pi-Sulfur interaction (orange dashed lines) seen between MET A:103 and the aromatic ring of the ligand. Pi-Pi T-Shaped interaction (pink dashed lines) is seen with PHE A:41 and the aromatic rings of the ligand. Pi-Alkyl interactions (blue dashed lines) seen with residues like ALA A:15, ILE A:16, and THR A:196 form such interactions with the aromatic rings of the ligand.
Figure 5: Two-dimensional interactions of 3-cinnamoyltribuloside against InhA.
Figure 7 shows a two-dimensional interaction diagram of Pyrazinamide bound to the pncA protein, highlighting several key interactions. Pyrazinamide engages in van der Waals interactions with multiple residues, including Valine (VAL) at positions 21 and 163, Isoleucine (ILE) at position 133, Leucine (LEU) at position 19.

Figure 7: Two-dimensional interactions of Pyrazinamide against pncA.
A novel plant-based secondary metabolite named 3-cinnamoyltribuloside has been identified as possessing anti-tuberculosis activity [40]. Molecular docking studies were conducted to compare its binding efficacy against five known anti-TB drugs: Isoniazid, Pyrazinamide, Bedaquiline, Pretomanid and Delamanid. These drugs, along with their respective targeted proteins were used as reference drugs. Blind docking was performed on both the control group and 3-cinnamoyltribuloside to explore potential binding sites on the entire protein. The PDB IDs of the drugs are as follows: Isoniazid (2nv6), Pyrazinamide (3pl1), Bedaquiline (5yio), Pretomanid (6g83) and Delamanid (6g83). Binding Affinity: The binding affinity (in kcal/mol) of 3-cinnamoyl tribuloside to the target protein was recorded. The docking simulation produced several binding poses for 3-cinnamoyl tribuloside within the active site of the target protein. Lower binding energy indicates a more stable ligand-protein complex. Initially, docking was carried out on one of the standard anti-tuberculosis drugs Isoniazid and natural product 3-cinnamoyltribuloside, revealing that the latter exhibited superior docking scores compared to Isoniazid. Consequently, further docking was conducted with another standard anti-tuberculosis drug Pyrazinamide, where 3-cinnamoyltribuloside demonstrated better scores. To further assess the potential of the natural component 3-cinnamoyltribuloside, docking studies were extended to three recently approved drugs by the United States Food and Drug Administration (US-FDA): Bedaquiline (2012), Delamanid(2014) and Pretomanid (2019). Figure 9 shows a two-dimensional interaction diagram of 3-cinnamoyl tribuloside bound to the pncA protein, highlighting various interactions between the ligand and amino acid residues. The interactions include van der Waals forces with several residues such as Serine (SER) at position 235, Aspartic Acid (ASP) at position 232, Isoleucine (ILE) at position 234, Arginine (ARG) at position 242, Phenylalanine (PHE) at positions 289 and 362, Glycine (GLY) at position 293, Tryptophan (TRP) at positions 230 and 296, Threonine (THR) at position 288, and Tyrosine (TYR) at position 297. Conventional hydrogen bonds are indicated by green dashed lines, formed with ASP 232, ARG 242, and SER 235. Carbon hydrogen bonds (light green dashed lines) are formed with TRP 230 and GLY 293. A pi-cation interaction (orange dashed line) is seen with ARG 242. Pi-sigma (pink dashed line), pi-pi stacked (light pink dashed line), and pi-pi T-shaped (magenta dashed line) interactions involve residues like TRP 296 and TYR 297. These interactions together highlight the binding affinity and specificity of 3-cinnamoyltribuloside to the pncA protein, contributing to its potential biological activity.

Figure 9: Two-dimensional interactions of 3-cinnamoyltribuloside against pncA.
Figure 11 shows a two-dimensional interaction diagram of Bedaquiline bound to the ATP synthase protein, highlighting several types of interactions between the drug and amino acid residues. Bedaquiline engages in van der Waals interactions with residues such as Glycine (GLY) at position 118 and Aspartic Acid (ASP) at position 48. It forms conventional hydrogen bonds with Arginine (ARG) at position 115 and Glutamic Acid (GLU) at position 31, shown with green dashed lines. Carbon hydrogen bonds, indicated by light green dashed lines, are also present with ARG 115. A significant pi-cation interaction with Arginine (ARG) at position 26 is marked by an orange dashed line. Additionally, Bedaquiline forms pi-sigma interactions with Alanine (ALA) at position 116 and pi-alkyl interactions with Phenylalanine (PHE) at position 24 and Isoleucine (ILE) at position 120, illustrated with pink and light pink dashed lines, respectively. There are also alkyl interactions with Valine (VAL) at position 51 and Alanine (ALA) at position 112.
Figure 11: Two-dimensional interactions of Bedaquiline against ATP Synthase.
Figure 13 shows shows a two-dimensional interaction diagram of 3-cinnamoyltribuloside binding to the ATP synthase enzyme, highlighting the various molecular interactions involved in this binding process. The interactions depicted include van der Waals forces (in green), conventional hydrogen bonds (blue), and pi-cation bonds (orange). Additionally, there are pi-anion interactions (red), pi-sigma interactions (purple), and pi-alkyl interactions (light purple), each playing a role in stabilizing the ligand within the enzyme's active site. Key residues involved in these interactions include ARG (arginine) at positions 62 and 115, which form pi-cation and pi-donor hydrogen bonds, respectively, indicating crucial points of interaction. Other significant interactions involve residues such as ASP (aspartic acid) at position 60, forming pi-anion interactions, and PHE (phenylalanine) at positions 22 and 24, engaging in van der Waals interactions. Additional residues like ALA (alanine) at positions 112 and 116, VAL (valine) at positions 51 and 53, and GLY (glycine) at positions 57 and 118 participate in a mix of pi-sigma and van der Waals interactions. These interactions collectively highlight the complex binding mechanism of 3-cinnamoyltribuloside to ATP synthase, emphasizing the multifaceted nature of the molecular interactions that facilitate its binding and potential inhibitory effects on the enzyme.

Figure 13: Three-dimensional interactions of 3-cinnamoyltribuloside against ATP Synthase.
Figure 15 shows two dimensional between Pretomanid against DprE1, it shows the van der Waals interactions, represented in green, involve numerous residues such as GLY B:133, HIS B:132, GLY B:117, THR B:118, PRO B:116, GLY B:57, SER B:59, ARG B:58, THR B:122, LEU B:56, GLY B:125, GLY B:55, CYS B:129, LYS B:418, ALA B:126, and TYR B:415. Conventional hydrogen bonds, indicated by green dashed lines, are formed with HIS B:132, GLY B:117, GLY B:57, SER B:59, and THR B:122. Carbon hydrogen bonds, depicted with light green dashed lines, are observed with ARG B:58 and VAL B:121. Halogen bonds (Fluorine), marked in cyan dashed lines, involve interactions with HIS B:132 and GLY B:117. Alkyl interactions, shown in light purple shading, occur with ALA B:417 and ILE B:131. Pi-alkyl interactions, highlighted in purple dashed lines, involve PRO B:116, ARG B:58, and VAL B:121.

Figure 15: Two-dimensional interactions of Pretomanid against DprE1.
One the other hand Figure 17 shows two dimensional interaction of 3-cinnamoyltribuloside against DprE1 shows Van der Waals interactions, represented in green, involve several residues, THR B:252, THR B:225, LEU B:250, SER B:195, ALA B:251, THR B:194, TYR B:226, THR B:192, PRO B:193, ASN B:135, SER B:138, ASN B:144, ARG B:147, ALA B:139, and LYS B:88. Conventional hydrogen bonds, depicted by green dashed lines, are formed with residues such as ASN B:144, ARG B:147, ALA B:251, THR(B:225, THR B:252, SER B:195, ASN B:135, and THR B:194. Pi-cation interactions, indicated by orange dashed lines, occur with ARG B:249 and HIS B:145. Pi-sigma interactions, shown in pink dashed lines, involve residues THR B:196 and PRO B:193. Pi-alkyl interactions, highlighted in purple shading, include interactions with residues TYR B:192 and HIS B:145. These diverse interactions collectively enhance the ligand's binding affinity and specificity to the protein, thereby contributing to the overall molecular stability and biological activity of the complex.

Figure 17: Two-dimensional interactions of 3-cinnamoyltribuloside against DprE1.
Figure 19 exhibits interaction between Delamanid against DprE1 Van der Waals interactions, shown in green, occur with residues GLY A:331, PHE A:332, GLY A:17, TRP A:16, GLY A:117, PRO A:316, VAL A:365, LYS A:134, and SER A:228. Conventional hydrogen bonds, represented by green dashed lines, are formed with residues GLY A:331, ASP A:389, GLY A:17, GLY A:117, and LYS A:134. Carbon hydrogen bonds, indicated by light green dashed lines, are seen with residues TYR A:60 and THR A:118. Pi-anion interactions, highlighted in orange dashed lines, involve ASP A:389. Pi-sigma interactions, depicted in pink dashed lines, are present with HIS A:315. Alkyl interactions, shown in light purple shading, include residues HIS A:315 and VAL A:365. Pi-alkyl interactions, represented by purple dashed lines, occur with TYR A:60, TYR A:314, and PRO A:316.

Figure 19: Two-dimensional interactions of Delamanid against DprE1.
Whereas Figure 21 shows last interaction between 3-cinnamoyltribuloside and it includes Van der Waals interactions with SER A:235, ASP A: 232, ILE A:234, GLY A: 293, and two residues of PHE A: 362 and PHE A: 289. Conventional hydrogen bonds are observed with TRP A: 230 and THR A: 288. There is a carbon hydrogen bond with TRP A:296. A pi-cation interaction is present with ARG A:242, providing an important electrostatic interaction. Pi-sigma interactions occur with TYR A:297, enhancing the binding through aromatic stacking. Additionally, pi-pi stacking interactions are observed with PHE A:289 and PHE A:362, which further stabilize the ligand through aromatic ring interactions. Lastly, a pi-pi T-shaped interaction is seen with TRP A:230, which contributes to the overall stability and binding affinity of the ligand.
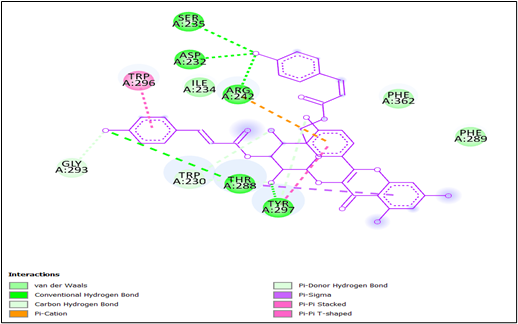
Figure 21: Two-dimensional interactions of 3-cinnamoyltribuloside against DprE1.
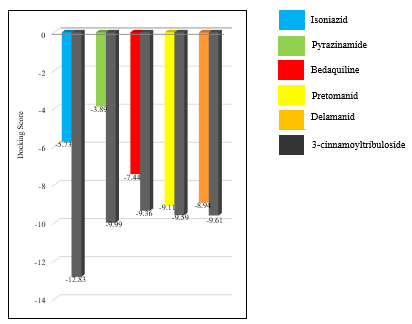
Comparison with Reference Ligands: The binding affinity and interaction profile of 3-cinnamoyl tribuloside were compared with known inhibitors of the target protein Isoniazid (2nv6), Pyrazinamide (3pl1), Bedaquiline (5yio), Pretomanid (6g83) and Delamanid (6g83) and to assess its relative efficacy. The docking score for InhA of isoniazid was found to be -5.73 whereas of 3-cinnamoyltribuloside was found to be -12.83 The docking score for pncA of pyrazinamide was found to be -3.89 whereas of 3-cinnamoyltribuloside was found to be -9.99. The docking score for ATP synthase of bedaquiline was found to be -7.44 whereas of 3-cinnamoyltribuloside was found to be -9.36. The docking score for DprE1 of pretomanid was found to be -9.11 and of delamanid was found to be -8.94 whereas of 3-cinnamoyltribuloside was found to be -9.59 and -9.61, respectively. The reference ligands, which have been experimentally validated, typically exhibited binding affinities in the range of -3.89 to -9.11 kcal/mol. The higher binding affinity of 3-cinnamoyl tribuloside suggests that it may be a more potent inhibitor. Additionally, the interaction profile of 3-cinnamoyl tribuloside was found to be similar to that of the reference ligands, with comparable hydrogen bonding and hydrophobic interactions. This further supports the potential of 3-cinnamoyl tribuloside as a bioactive compound capable of effectively interacting with the target protein. Structural Analysis: The structural analysis of the ligand-protein complex revealed that 3-cinnamoyl tribuloside adopts a conformation that allows optimal interactions with the active site residues. The flexibility of the cinnamoyl group enables it to fit snugly within the binding pocket, maximizing contact with the protein. This structural adaptability is likely a key factor in the ligand's high binding affinity and specificity. Potential Biological Implications: The strong binding affinity and favorable interaction profile of 3-cinnamoyl tribuloside suggest that it may have significant biological activity. The ability to form stable hydrogen bonds and hydrophobic interactions with the target protein indicates that it could effectively inhibit the protein's activity, potentially leading to therapeutic effects. Table 1 represents Docking score of reference drugs and 3-cinnamoyltribuloside against target proteins Further experimental validation, including in vitro and in vivo studies, is necessary to confirm these findings and explore the compound's pharmacological potential.



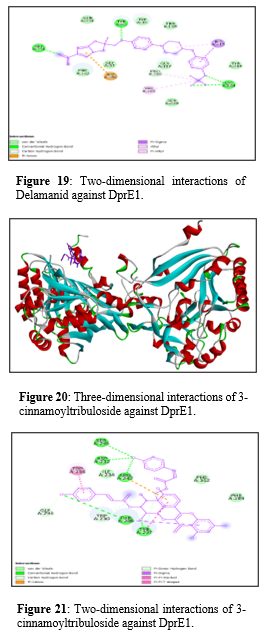
Table 1: Docking score of reference drugs and 3-cinnamoyltribuloside against target proteins
|
Target |
Target Protein PDB ID |
Reference |
Docking |
Experimental |
Docking Score |
|
InhA |
2NV6 |
Isoniazid |
-5.73 |
3-cinnamoyltribuloside |
-12.83 |
|
pncA |
3PL1 |
Pyrazinamide |
-3.89 |
-9.99 |
|
|
ATP Synthase |
5YIO |
Bedaquiline |
-7.44 |
-9.36 |
|
|
DprE1 |
6G83 |
Pretomanid |
-9.11 |
-9.59 |
|
|
DprE1 |
6G83 |
Delamanid |
-8.94 |
-9.61 |
CONCLUSION
The results of our study interpreted that the natural compound, 3-cinnamoyltribuloside had exhibited best docking score against two of the first line anti-TB drugs Isoniazid and Pyrazinamide and also against the recently approved US-FDA anti-TB drugs Bedaquiline, Delamanid and Pretomanid. Among which the docking score against Isoniazid target, InhA is the finest of all. The research is clearly showing that the 3-cinnamoyltribuloside can inhibit targets of the above mentioned drugs namely, InhA, pncA, ATP Synthase, DprE1, DprE1, respectively. Based on the analysis, it can be inferred that our control drug (3-CT) showed most favourable docking scores among the reference drugs Bedaquiline, Pretomanid, Delamanid, Pyrazinamide and Isoniazid. In conclusion, the molecular docking study of 3-cinnamoyl tribuloside demonstrated strong binding affinity and favorable interaction profile with the target protein. The presence of multiple hydrogen bonds, hydrophobic interactions, and π-π stacking interactions suggests that 3-cinnamoyl tribuloside can effectively bind to and potentially inhibit the target protein. These findings provide a solid foundation for further experimental investigations into the therapeutic potential of 3-cinnamoyl tribuloside. These in-silico predictions provide a valuable starting point for drug discovery efforts, but the ultimate determination of efficacy and therapeutic potential requires rigorous experimental validation in biological systems.
LIST OF ABBREVIATIONS
TB: Tuberculosis
Mtb: Mycobacterium tuberculosis
MM: Mycobacterium madagascariense
MIP: Mycobacterium indicus pranii
WHO: World Health Organization
FDA: Food Drug Administration
USFDA: United States Food Drug Administration
PZA: Pyrazinamide
INH: Isoniazid
PDB: Protein Data Bank
PDBQT: Protein Data Bank Quasi-Biennial Triennial
GPF: Grid Parameter File
DPF: Docking Parameter File
GLG: Grid Log File
DLG: docking log file
MGL: Molecular Graphics Laboratory
OAT: Organic Anion Transporter
Ddn: Deazaflavin-dependent Nitroreductase
MICs: Minimum Inhibitory Concentrations
EMA: European Medicines Agency
MDR-TB: Multidrug-Resistant Tuberculosis
XDR-TB: Extensively Drug-Resistant Tuberculosis
InhA: Inhibin Subunit Alpha
PncA: Pyrazinoic Acid
DprE1: Decaprenyl phosphoryl-β-d-ribose 2'-epimerase
ATP: Adenosine Triphosphate
AVAILABILITY OF DATA AND MATERIALS
The data supporting the findings of the article is available within the article.
FUNDING
This research received no external funding.
CONFLICT OF INTERESTS
No conflicts of interest were reported by the authors.
REFERENCE


Dr. Komal Govind Sangu*, Ishwari Tukaram Katre, Dhirajkumar Rajkumar Gupta, Molecular Docking Studies Of 3-Cinnamoyltribuloside, Int. J. of Pharm. Sci., 2025, Vol 3, Issue 9, 3223-3248 https://doi.org/10.5281/zenodo.17222365
 10.5281/zenodo.17222365
10.5281/zenodo.17222365