
We use cookies to ensure our website works properly and to personalise your experience. Cookies policy
College of Pharmaceutical Sciences, Government Medical College, Thiruvananthapuram, 695011.
Quinazoline and quinazolinone containing compounds are important class of nitrogen containing heterocyclic compounds having diverse therapeutic activities. A variety of marketed drugs containing these moieties are available now and these compounds have been reported to exhibit a wide spectrum of biological functions. Their synthetic feasibility and versatility in structural alterations and fictionalization adds their allure in medicinal chemistry. It is interesting that the anticancer activity of these compounds is more projected. However, their activity is not only restricted to single cancer target. Therefore, it is very interesting to explore quinazoline and quinazolinone based compounds having immense therapeutic potential on multiple cancer targets. A recent update on various quinazoline derivatives acting on different molecular targets for cancer treatment has been combined in this review.
Heterocyclic compounds are often found to have interesting applications in different fields of medicinal chemistry. Especially nitrogen containing heterocycles are important class of compounds and their significance is well established.3 Most of the currently available drugs contain heterocyclic moieties and nitrogen containing heterocycles are integral part of numerous natural products such as vitamins, hormones, antibiotics, alkaloids, herbicides etc.1
Chemistry of Quinazoline and Quinazolinone
Quinazoline and quinazolinone are nitrogencontaining heterocyclic compounds having a benzene ring fused with a pyrimidine ring. So it can be called as Benzopyrimidine. The synthesis of quinazoline was first reported in 1895 by August Bischler and Lang. In 1903, Siegmund Gabriel reported the synthesis of parent quinazoline from o-nitrobenzylamine and conducted extensive studies on quinazoline.1
Quinazoline is also called 1,3-diazanaphthalene because of being an aza derivative of quinoline. Depending on the varying positions of nitrogen atoms, quinazoline ring is isomeric with other diazanaphthalenes of benzodiazine subgroups, i.e.cinnoline, quinoxaline, and phthalazine.2
Quinazolinone is an extension of quinazoline further fitted with a carbonyl group. The first reported synthesis of quinazolinone was in 1989 by P. Gries and is known as Griess synthesis. Based on the different positions of the carbonyl group, two isomers are possible for quinazolinone, i.e. 2(1H)quinazolinone and 4(1H)quinazolinone, with the 4-isomer is being more common. A dicarbonyl form, 2,4(1H,3H)quinazolinedione is also known to exist.2
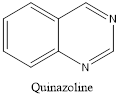
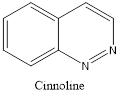
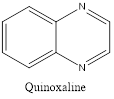
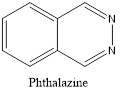
Isomers of Quinazoline
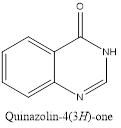
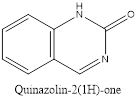
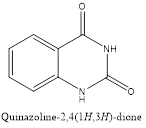
Isomers of Quinazolinone
Currently, cancer is a severe health problem with boundless predominance. It is a complex disease characterized by uncontrolled cellular division without proper differentiation which results in abnormal cells having the potential to invade to other parts of the body. Most of the available anticancer drugs are cytotoxic, unselective and ultimately results in resistance. So there has always been a focus on developing safer and targeted anticancer drugs to treat increasing number of patients with disseminated malignancies.4,5
Numerous established anticancer quinazoline and quinazolinone derivatives form a new class of chemotherapeutic agents which are found to act by inhibiting various protein kinases and other molecular targets.
Table 1. Various quinazoline and quinazolinone based anticancer drugs.
|
Name |
Structure |
Specific Target |
|
Quinazoline derivatives |
||
|
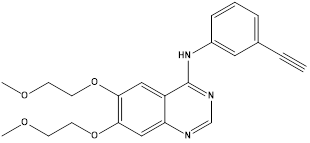
Erlotinib.6 |
|
EGFR inhibitor (Tyrosine kinase enzyme) |
|
Gefitinib.7 |
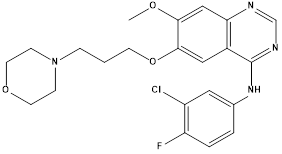
|
EGFR inhibitor (Tyrosine kinase enzyme) |
|
Lapatinib.8 |
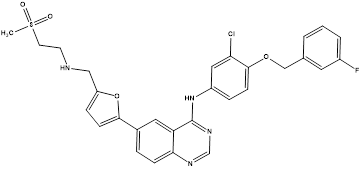
|
Dual tyrosine kinase enzyme inhibitor (EGFR and HER-2) |
|
Afatinib |
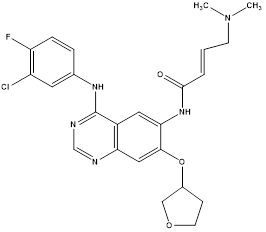
|
ErbB family of tyrosine kinase inhibitor |
|
Vandetanib |
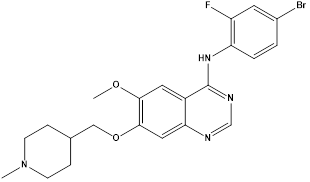
|
VEGFR AND EGFR Inhibitor |
|
Cediranib |
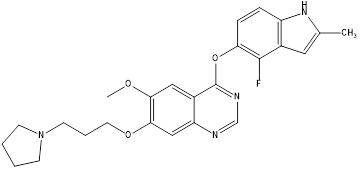
|
VEGFR |
|
Quinazolinone derivative
Raltitrexed.9 |
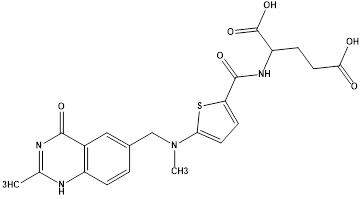
|
Thymidylatesynthase inhibitor |
ANTICANCER POTENTIAL OF QUINAZOLINE AND QUINAZOLINONE DERIVATIVES
Quinazoline and quinazolinone derivatives comprise an exceptional class of anticancer agents active against various tumors through different targets. Most of the quinazoline analogues primarily act by inhibiting different protein kinases which ultimately inhibit the replication and transcription of DNA and thereby impede the tumor growth.10,11
Novel quinazolinone derivatives as potential anticancer agents via PI3K inhibition
PI3K/Akt pathway have been extensively studied as one of the critical pathways regulating cellular processes like cell survival, proliferation, differentiation, migration. This signal transduction system is activated by a wide range of receptor tyrosine kinases, generates an important second messenger phosphatidylinositol-3,4,5-triphosphate (PIP3) which regulates cellular functions by phosphorylating downstream effectors. It is the most commonly activated signal transduction system in cancer that links oncogenes and multiple receptor classes to many important cellular functions. Any abnormality in the activation of PI3K/Akt pathway is identified in most of the human malignant tumors make it a promising drug target for cancer therapy.12
Fifteen 6-(pyridine-3-yl) quinazolin-4(3H)-one derivatives were designed and synthesized by Huarong et al. Anticancer activities of synthesized compounds were evaluated and the potential mechanisms were explored. Several compounds showed antiproliferative activity against tested cancer cells including human non-small cell lung cancer (NSCLC) HCC827, human neuroblastoma SH-SY5Y and hepatocellular carcinoma LM3 cells. Among them, compound A and B showed best inhibitory activity against all the cancer cell lines and more active against HCC827 cells with IC50 values of 1.12μM and 1.20μM, respectively. In addition, A and B showed lower inhibitory activity against H7702 cells (human normal liver cells) with IC50 values of 8.66μMand 10.89Μm respectively, nearly 8-fold lower than that in HCC827 cells. These results suggested that compounds A and B had certain selectivity to tumor cells, compared to human normal cells. Since compound A showed best inhibitory activity, it was selected for further mechanism study.
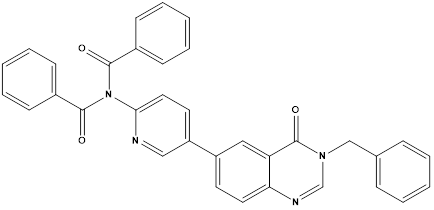
Compound A
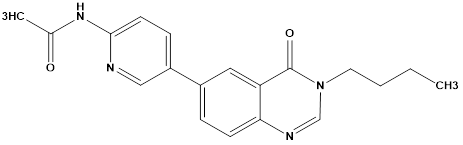
Compound B
Analyzed protein alterations of the critical components of PI3K/Akt pathway including p-Akt, p-GSK3β and p-mTOR using western blot to explore the effects of A at various dose on PI3/Akt pathway which showed that the phosphorylated levels of all these downstream targets were completely suppressed, indicating the inhibition of PI3K/Akt pathway. To confirm this, PI3K Kinase assay was performed using assay kit. Compound A markedly inhibited total PI3K kinase activities with IC50 values of 0.072μM.
Cell cycle distribution of compound A was examined, A induced G2 cell cycle arrest in a concentration dependent manner, and G1 phase was relatively decreased compared to that of control groups. Further elucidated the relation of cell cycle arrest to cell cycle checkpoint proteins, the expression of G2 cell cycle regulatory proteins was determined by western blot analysis. Reduced levels of CHK-1, p-CHK1, c-Myc and Cyclin B1 were observed on treatment with compound A.
Efficacy of cell apoptosis in HCC827 cells were assessed by examining the expression levels of apoptosis associated proteins by western blot analysis. The pro-apoptotic proteins cleaved- PARP, cleaved -caspase9 and BAX were upregulated by A treatment in a concentration dependent manner, but downregulated anti-apoptotic protein Bcl-2 which suggested that A induced apoptotic cell death via mitochondrial pathway in HCC827 cells.13
Collectively the results suggested that compound A might be a hit compound inhibitor of PI3K/Akt pathway.
Quinazoline based CDK2 inhibitors
Cyclin-dependent kinases (CDKs) belong to family of serine-threonine protein kinases, which is involved in regulation of cell cycle progression that uses ATP as a phosphate donor and carry out protein phosphorylation. They also regulate transcription, mRNA processing, differentiation of nerve cells and metabolism.14 Cyclin, a regulatory protein binds to CDK and form cyclin- CDK complex which phosphorylates substrates appropriately for the particular phase of cell cycle.15 CDKs are overactive or CDK inhibiting proteins are not functional in many types of cancers.16 Therefore, CDKs are most promising anticancer targets. Extensive efforts are done to develop quinazoline based CDK inhibitors.X-ray crystallographic analysis of anilinoquinazolines revealed the interaction of nitrogen atom at position 1 and aromatic hydrogen atoms at C2 and C8 of the quinazoline core with peptide strand of NH and carbonyl oxygen atoms of CDK2 enzyme.17
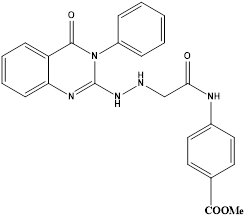
Eman R Mohammed et al synthesized and evaluated new quinazoline based compounds for their anticancer activity against sixty tumour cell lines. Compounds C and D showed excellent growth inhibition against the melanoma cell line MDA-MB-435 with GI% of 94.53 and 94.15, respectively. Cell cycle analysis showed that compound C led to cell cycle cessation at S phase and G2/M phase revealing that CDK2 could be the plausible biological target. Thus, the most cytotoxic candidates C and D were evaluated in vitro for their CDK2 inhibitory activity and were able to display significant inhibitory action.
The quinazolinones C and D displayed excellent growth inhibition against the melanoma cell line MDA-MB-435 (GI% ¼ 94.53 and 94.15, respectively). The quinazolinone C also exhibited promising activity against the glioblastoma cell line SNB-75 (GI% ¼ 63.09), while D showed potent to moderate activity against leukaemia cell lines SR (GI% ¼ 69.55) and K-562 (GI% ¼ 67.80). Compound C has emerged as the most active member against MDA-MB-435 with IC50 value of 3.03 mM which is 1.7-folds more potent than roscovitine, the reference standard (IC50 ¼ 5.26 mM). Compound D also showed good potency on MDA-MB-435 cell line with IC50 value of 6.22 mM which is comparable to that of the reference drug. The most active candidates C and D may serve as useful lead compounds in the search for promising anti-melanoma agents acting through CDK2 inhibition.18
Compound C
Compound D
Quinazoline based VEGFR-2 inhibitors
Vascular Endothelial Growth Factor Receptor(VEGFR) is associated with angiogenesis, a process of formation of new blood vessels from pre-existing one (neovascularization) which is essential for various physiological proceesses.19 It is mediated by vascular endothelial growth factor(VEGF) and is prerequisite for the rapid growth of the tumors. VEGF binds mainly to VEGFR-2 tyrosine kinase, activates the receptors by autophosphorylation.20 Overexpression of this VEGF is associated with various increased tumor vessel permeability, cancer recurrence etc. Therefore, VEGFR inhibition is one of the most significant targets in anticancer therapy.21
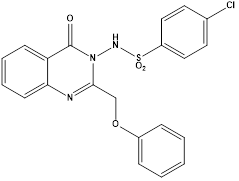
In search of selective VEGFR- 2 inhibitors, Abdallah E Abdallah et al synthesized new nineteen quinazolinone derivatives and biologically evaluated for their potential anticancer activity. All target compounds were evaluated in vitro for VEGFR-2 tyrosine kinase inhibition. Then, nine compounds of best results were further investigated by in vitro assay against three human cancer cell lines, namely HepG2, PC3 and MCF. N’ -{2-](3-Ethyl-6-nitro-4-oxo-3,4-dihydroquinazoline-2-yl)thio[acetyl}benzohydrazide (E) was found to be the most potent candidate as it showed IC50 = 4.6 ± 0.06 µM against VEGFR-2 kinase. It also exhibited IC50 = 17.23 ± 1.5, 26.10 ± 2.2 and 30.85 ± 2.3 µg/mL against HepG2, PC3 and MCF, respectively. At the same time it showed IC50 = 145.93 ± 1.1 µg/mL against the normal human lung fibroblasts cell line (WI-38), indicating good selectivity index. Further investigation into HepG2 cell cycle showed the ability of compound E to induce apoptosis and arrest cell growth at G2/M phase.22
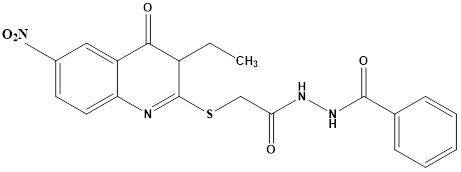
Compound E
Quinazoline based PARP-1 inhibitors
Poly (ADP-RIBOSE) Polymerase (PARP) is an enzyme which detects chemically or metabolically induced single stranded DNA breaks(SSB) and initiates immediate cellular response resulting in SSB repair.23 PARP-1 and PARP-32 are two forms of these enzymes. Despite of its importance I genomic repair mechanism, the substantial activation of PARP results in depletion of NAD+ and extended ADP ribosylation. In order to resynthesize NAD+, cellular depletion of ATP occurs which ultimately causes mitochondrial dysfunction and cell death.24 Thus, PARP inhibition is considered as a promising anticancer drug target as it plays an important role in programmed cell death and maintaining DNA integrity.
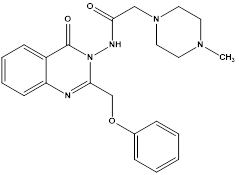
Sayed K. Ramadan et al reported an eco-friendly synthesis of a new series of quinazolinone-based derivatives as potential PARP-1 inhibitors. The 4-quinazolinone scaffold was utilized as a bioisostere to the phthalazinone core of the reference compound Olaparib. Most of the synthesized compounds displayed appreciable inhibitory activity against PARP-1. Compound G showed inhibitory activity at IC50 ¼ 30.38 nM comparable to Olaparib, which has IC50 ¼ 27.89 nM. Cell cycle analysis was performed for compounds Gand F, and both exhibited cell growth arrest at G2/M phase in the MCF-7 cell line. In addition, both compounds increased the programmed apoptosis compared to the control.
Compound F
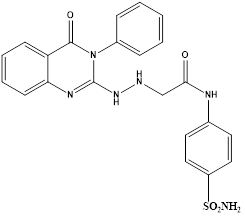
Compound G
The results of apoptosis assay revealed that compound F induced a total apoptotic effect equal 26.55% and G showed an effect equal to 19.31%, which were more than the control (1.57%) and comparable to that of Stauroporine. In detail, compounds F and G obviously induced early apoptosis by 3.47% and 6.11%, respectively, and they enhanced late apoptosis by 13.8% and 10.15%, respectively, when compared with the untreated control MCF-7 cells (0.61% and 0.11%, respectively).25
Quinazoline based inhibitors of breast cancer resistance proteins(ABCG2)
The ATP-binding cassette (ABC) transporter breast cancer resistance protein (BCRP), sometimes referred to as ABCG2, is known to contribute to multidrug resistance (MDR) in a variety of cancer cells. The ability of ABC transporters to extrude medications from malignant cells is the cause of this resistance. An increased amount of endogenous and exogenous chemicals are actively effluxed across the cell membrane by BCRP overexpression. From a physiological standpoint, BCRP removes hazardous and poisonous foreign compounds via the biliary system, gut, blood-brain, placental, and potentially blood-testis barriers.26 Drugs can concentrate in cells and exhibit their anticancer effects via blocking BCRP. As a result, co-administration of BCRP inhibitors and anticancer medications is an intriguing strategy to address the issue of MDR caused by ABC transporters.27There have been reports of several quinazoline-based drugs having impressive BCRP inhibitory properties.
Chao-Yun Cai et alreported synthesis of a set of twenty-two quinazolinamine derivatives were developed, demonstrating strong inhibitory effects on BCRP (breast cancer resistance protein). Quinazolinamine,H that contained cyclopropyl was found to be an inhibitor of BCRP, and quinazolinamineI that contained azide also demonstrated BCRP inhibitory action. Compound H caused a change in the location of P-gp and BCRP in cells, which prevented the two ATP-binding cassette (ABC) transporters from effluxing anticancer medications. Furthermore, it was shown that both H and I exhibited a considerable increase in the ATP hydrolysis of the BCRP transporter. This suggests that they may function as competitive substrates of the BCRP transporter, leading to an increase in the accumulation of mitoxantrone in H460/MX20 cells that overexpress BCRP. Azide derivative I showed a stronger inhibitory effect on BCRP following UV activation and can be a useful probe for examining quinazolinamine derivatives using BCRP. Notably, when compared to Ko143, the dual BCRP and P-gp inhibitors H, and BCRP inhibitor I demonstrated improved metabolic stability.28
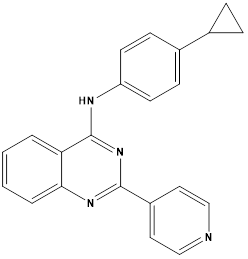
Compound H
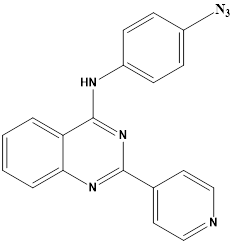
Compound I
N-aryl sulphonamide quinazoline derivatives acting on Hippo signalling pathway
The extremely conserved Hippo signaling system was first discovered in Drosophila.[29-31]In recent years, it has been discovered to be a critical regulator of cell survival, proliferation, and differentiation in mammals.[32-37] The Hippo pathway in mammalian cells is mediated by protein kinases of Salvador homolog (SAV) 1, Mammalian STE20-like protein kinase (MST) 1/2, Large Tumor Suppressor (LATS) 1/2, and Mps one binder kinase activator-like (MOB) 1. These proteins control transcriptional enhanced associate domain (TEAD), transcriptional co-activator with PDZ-binding motif (TAZ), and downstream effectors of yes-associated protein (YAP).[29-33]Phosphorylated MST1/2 has the ability to phosphorylate and activate LATS1/2 in the signaling cascade. In the cytosol, phosphorylated LATS1/2 would phosphorylate YAP. MST1/2 may activate the scaffold proteins MOB1 and SAV1. The ubiquitin-proteasome would be responsible for breaking down the phosphorylated YAP.38Non-phosphorylated, activated YAP would move from the nuclear cytoplasm of the cell.39,40 In cell nucleus, YAP and TEADs work cooperatively to control the target oncogene proteins, such as c-Myc and Bcl-2. Many cancers including breast cancer,41 lung cancer,42 colorectal cancer,43 stomach cancer,44 and prostate cancer,45 have an over-activation of YAP, and the expression of its negatively regulated oncogenic target gene is higher than that of normal cells.46 Because Hippo/YAP has a direct relationship with tumor formation, it is currently thought to be a prospective target for cancer therapy.[47-49]But there aren't many known molecules that alter the Hippo pathway, and the majority of them don't have strong anticancer effects.
Jin-Bo Niu et al discovered compound J, a derivative of N-aryl sulphonamide and quinazoline, as an anti-gastric cancer agent. It inhibited YAP activity by activating p-LATS and demonstrated strong antiproliferative ability, with IC50 values of 0.36 lM (MGC-803 cells), 0.70 lM (HCT116 cells), 1.04 lM (PC-3 cells), and 0.81 lM (MCF-7 cells), respectively. Compound J dramatically down-regulated the expression of YAP in vivo and was efficient in inhibiting the formation of MGC-803 xenograft tumours in naked mice without showing any signs of toxicity. Compound J caused intrinsic apoptosis, prevented the formation of cell colonies in MGC-803 and SGC-7901 cells, and stopped cells in the G2/M phase. As a result, compound J is to be regarded as an anti-gastric cancer drug that acts by stimulating the Hippo signaling system, which may facilitate the development of aa novel cancer therapy approach.50
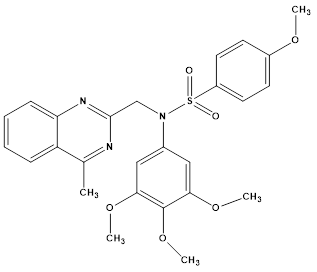
Compound J
Quinazolinone based HDAC6 inhibitors
The enzymes that regulate intracellular histone acetylation include histone deacetylases (HDACs) and histone acetyl transferases (HATs). HATs act as "writers," moving the acetyl group from acetyl-CoA to the amine group of lysine residues on histones and non-histone proteins. This process broadens the landscape of the chromosome so that regulatory proteins or appropriate transcription factors can detect it. Later, transcription factors are recruited for the expression of the resulting gene.[51-56] The creation of isozyme-selective HDAC inhibitors with lower side effects and higher potency is therefore given top focus in research. Class II enzyme histone deacetylase 6 (HDAC6) is a member of the class IIb group. It is mostly located in the cytoplasm and is distinct from other HDACs in both structure and function. HDAC6, one of the largest members of the HDAC family, with a total length of 1255 amino acids. Viral infection, immunological synapse, and microtubule-dependent cell motility are all linked to HDAC6.57,58 HDAC6 has a major impact on a number of tumorigenesis-related processes, such as enhanced cancer cell invasion, migration, and proliferation.[59-61]
It's interesting to note that HDAC6 inhibitors have immunomodulatory qualities as well, suggesting that they could be employed as therapeutic agents in cancer immunotherapy.62 The expression of the immunosuppressive protein PD-L1 was decreased in melanoma tumor cells upon suppression of HDAC6. By affecting STAT3 recruitment and activation, this impact was accomplished.63 Targeting HDAC6 specifically is intended to optimize the pharmacological effects while reducing the negative effects associated with pan-HDAC inhibitors because HDAC6 differs from the other HDAC isozymes in both structure and function.64.65 This theory also encouraged the development of novel HDAC6-selective inhibitors.
Using novel quinazoline-4-(3H)-one as the cap group and benzhydroxamic acid as the linker and metal-binding group,a series of novel quinazoline-4-(3H)-one derivatives were designed and synthesized as histone deacetylase 6 (HDAC6) inhibitors by Yogesh Mahadu Khetmaliset al. Nineteen unique quinazoline-4-(3H)-one analogues in total were discovered. The target compounds' structures were identified using elemental analysis, LC-MS, 13C-NMR, and 1H-NMR. The compounds that were characterized were examined for their ability to inhibit HDAC8 class I, HDAC4 class IIa, and HDAC6 class IIb. With an IC50 value of 150 nM, compound K was shown to be the most effective and selective HDAC6 inhibitor among those tested. Several tumor cell lines (MCF7, B16, and HCT116) shown strong antiproliferative activity in response to some of these drugs. Out of all the compounds tested for their anticancer effect against cancer cell lines, compound L was found to be the most active with an IC50 of 13.7 µM; it also demonstrated cell-cycle arrest in the G2 phase and promoted apoptosis. Moreover, we observed a significant decrease in the ability of cancer cells to form colonies when compound L was present. At the intracellular level, compound L was found to selectively inhibit HDAC6 by monitoring the acetylation of α-tubulin, with a limited effect on acetyl-H3. Notably, the obtained results suggested a potent effect of compound L at sub-micromolar concentrations as HDAC6 inhibitors in vitro.66
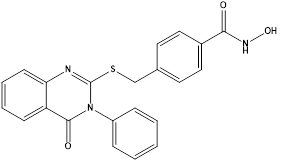
Compound K
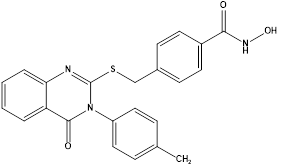
Compound L
FUTURE ASPECTS
Despite the fact that different quinazoline derivatives have been developed as anticancer medicines, a number of obstacles are pushing the boundaries of research for the next generation of small-molecule kinase inhibitors based on quinazoline. Even if there have been developments recently to create targeted anticancer drugs, yet a good selectivity profile is still required to strike a compromise between toxicity and efficacy. As therapeutically beneficial chemotherapeutic drugs, several kinase inhibitors with significant promise for the treatment of CNS and cardiovascular diseases may also be investigated.
CONCLUSION
After a phase of nonselective harmful medications, the research on innovative cancer treatments has moved on to less toxic selective agents. One of the main problems with the chemotherapeutic medicines that are currently on the market is their lack of selectivity, which can lead to toxicity. Consequently, there is strong evidence to support the use of targeted therapy with site-specific action in the management of this fatal illness. The quinazoline pharmacophore has been extensively explored for the treatment of cancer as a consequence of extensive research efforts in the field of drug discovery.
Quinazolines have been developed for kinase-targeted cancer therapy in recent decades due to advances in our understanding of cancer biology. Numerous literature reports are adding to the vast body of scientific evidence demonstrating the therapeutic potential of quinazoline-based anticancer agents. Several molecules from this diverseclass of drugs acting through novel targets have been approved by FDA and are available for clinical use against advanced cancer treatment. Most of the modifications carried out at 4-,6- and 7- positions of the quinazoline skeleton to develop anticancer molecules resulted in compounds
with tremendous biological activity against cancer. Small structural changes in the
quinazoline framework bring about dramatic changes in the potency and specificity of
resultant analogues towards various molecular targets of cancer therapy. Irrespective of the
availability of various quinazoline based clinical candidates, enough potential remains in this
privileged scaffold to be explored for producing better therapeutic agents. Further structural
modifications in quinazoline nucleus might lead to newer derivatives with potency more than
available drugs. This study provides scientists working toward the development of novel target-specific therapeutic candidates for cancer therapy with fresh insights and enhanced comprehension.
REFERENCES

Airin Benny, Aravind Ayyappan, Krishna Priya A, Quinazoline Based Molecular Hybrids as Potential Therapeutic Agents in Tumour Therapy, Int. J. of Pharm. Sci., 2025, Vol 3, Issue 9, 2447-2461. https://doi.org/10.5281/zenodo.17175617
 10.5281/zenodo.17175617
10.5281/zenodo.17175617