
We use cookies to ensure our website works properly and to personalise your experience. Cookies policy
Shivaji University, Kolhapur.
Since isoxazole derivatives have a variety of pharmacological actions, such as antibacterial, anticancer, and anti-inflammatory qualities, they are frequently investigated in drug discovery. We created and assessed new isoxazole derivatives in this work based on their molecular interactions with a chosen therapeutic target and pharmacokinetic characteristics. To use computational methods to evaluate novel isoxazole derivatives' drug-likeness, binding affinity, and ADMET (Absorption, Distribution, Metabolism, Excretion, and Toxicity) characteristics.
As a five-membered oxygen–nitrogen heterocycle, isoxazole is a privileged scaffold in medicinal chemistry, exhibiting a variety of biological activities such as antimicrobial, anticancer, anti-inflammatory, anticonvulsant, and analgesic properties. The existence of the isoxazole ring leads to improved bioactivity, metabolic stability, and favorable pharmacokinetic profiles, making it a valuable core structure for novel drug development. Heterocyclic compounds are essential in drug discovery, and isoxazole derivatives are receiving a lot of attention because of their wide range of pharmacological applications. Given the therapeutic importance of isoxazole derivatives, this study intends to design and assess novel isoxazole-based compounds using molecular docking and in silico ADMET screening. The main goal is to find derivatives that have strong binding affinities toward a chosen target protein linked to a particular disease and optimal pharmacokinetic properties. The results of this study will be used to prioritize the most promising candidates for additional in vitro and in vivo validation, opening the door for possible drug development. According to the American Cancer Society's Global Cancer Statistics 2024 report, 9.7 million people worldwide lost their lives to cancer in 2022, and about 20 million new cases of cancer were diagnosed. It is estimated that 35 million cases of cancer will occur by 2050. Results from the Trail Assigning Individualized Options for Treatment (Rx) clinical trial show that chemotherapy after surgery is not beneficial for the majority of women with early-stage breast cancer. This information was acquired in 2018. 2018: Larotrectinib received accelerated FDA approval to treat solid tumors with the NTRK gene that locally evolved or metastatic in both adult and pediatric patients. In 2022, there had been an projected 20 million cancer deaths worldwide, and the disease was responsible for almost 10 million deaths.
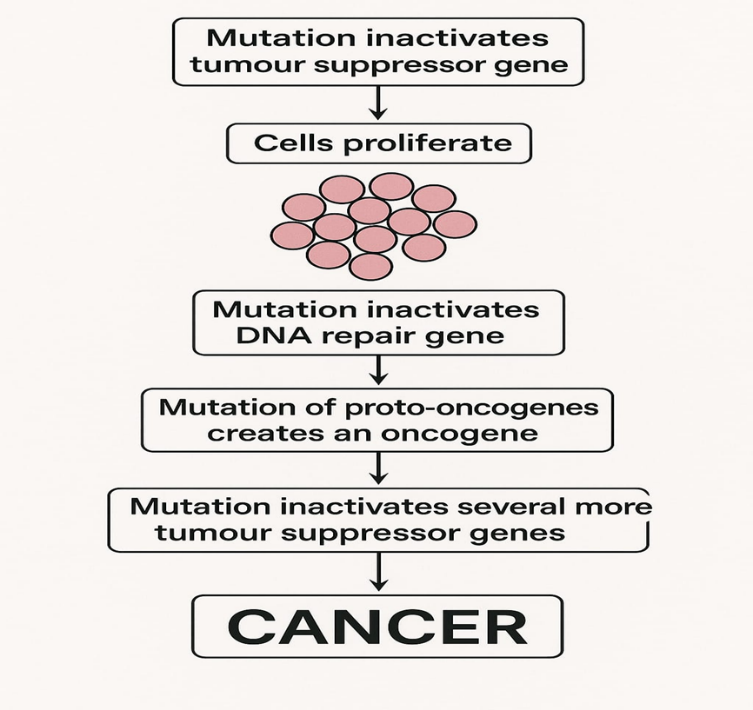
Figure no 1: Mechanism of cancer formation
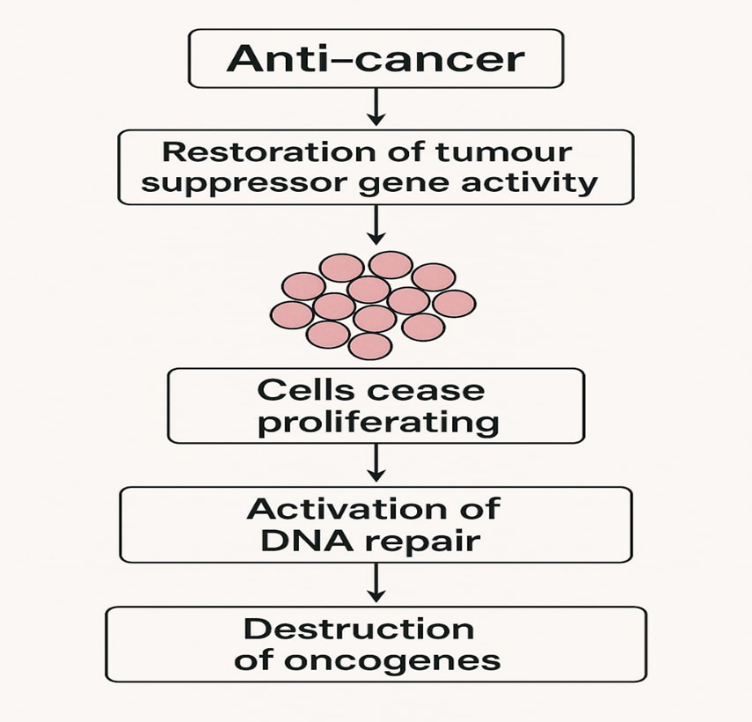
Figure no 2: Mechanism of action of anticancer drug.
MATERIAL AND METHOD:
Selection and Design of Novel Derivatives:
Computational drug design techniques were used to create a number of new compounds based on the isoxazole scaffold. CHEMDRAW was used to create and optimize the molecular structures. Using PyRx software , all generated 2D or 3D structures converted into PDB format for subsequent applications in automated docking and molecular cleaning studies.
Protein Selection and Preparation:
The study target was selected based on Swiss Target Prediction Report. The three- dimensional crystallography design of Protease inhibitors was retrived from PDB, with water molecules and hetero atoms removed. The BIOVIA was utilized to examine the operating pocket and understand the dispersion of amino acid residues, which are illustrated in the companying figure. Additionally, the protein structure was plotted on a Ramachandran plot.
Predictions of Pharmacokinetics (ADME) toxicology:
Various in silico methods were used to assess the pharmacokinetic and toxicity profiles of the prepared derivatives: Utilizing SwissADME and Molinspiration (Lipinski's Rule of Five), the drug's likeness and physicochemical properties were determined. These tools compute physicochemical properties, lipophilicity, water solubility, and medicinal chemistry friendliness. The In-Silico ADME / Tox profile is a useful tool to predict the pharmacological properties of drug candidates, especially in preclinical stages.
Bioactive molecules drug likeness score:
Compounds drug similarity can be predicted using online servers such as MOLINSPIRATION, SWISS ADME which analyze various molecular properties. Molinspiration utilizes cheminformatics tools to evaluate the bioactivity of compounds based on their chemical structures, focusing on parameters like h bond donors and acceptors, molecular weight and log p values.
Molecular Docking Procedure:
1. Ligand Preparation.
Ligand structures were extracted from the PubChem database in SDF format. The chemicals were imported into Open Babel, which is incorporated into PyRx and energy was minimized using the Universal Force Field (UFF) with 200 conjugate gradient minimization steps. The reduced structures were saved as PDBQT files for docking.
2. Target Protein Preparation
The target protein's three-dimensional (3D) structure was retrieved from the RCSB Protein Data Bank (PDB). Water molecules and non-standard residues (such as co-crystallized ligands and ions) were eliminated with the Discovery Studio Visualizer (BIOVIA). Polar hydrogens were introduced, and Gasteiger charges were assigned in PyRx. The cleaned protein structure was converted to PDBQT format using PyRx's inbuilt AutoDock Tools.
3. Molecular docking
In PyRx, ligands were docked into the target protein's active site with AutoDock Vina. The software created several conformations of each ligand and reported the binding affinity (ΔG, in kcal/mol) for each position. The position with the lowest binding energy was chosen for further research.
4. Post-Docking Analysis
The resulting docking poses were visualized and evaluated with Discovery Studio Visualizer. Protein-ligand interactions were investigated, including:
To identify important binding residues, 2D interaction diagrams and 3D binding poses were produced for the best-docked conformations.
RESULT AND DISCUSSION:
The goal of the study was to evaluate the drug like properties of new compounds and assess their binding affinity for 1PK4. Novel derivatives obtain from Insilco design were assessed for their ADMET properties and potential anticancer activity. The ADMET parameters were predicted using tools like Swiss ADME and Molinspiration, and all measured values were compared with those of standard compound.
Table no1: 2D structure of derived compounds.
|
Compound |
STRUCTURE |
Compound |
STRUCTURE |
|
1 |
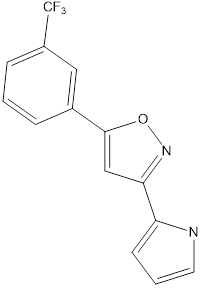
|
2 |
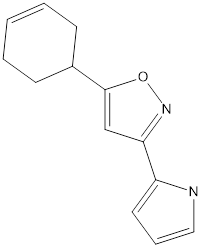
|
|
3 |
4 |
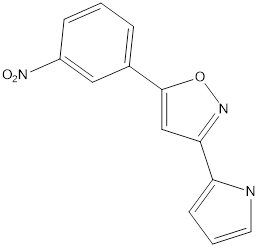
|
|
|
5 |
|
6 |
|
|
7 |
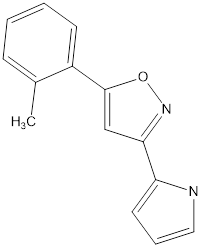
|
8 |
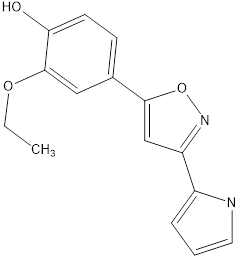
|
|
9 |
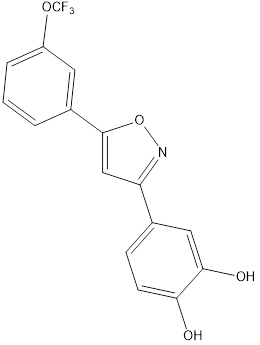
|
10 |
|
Table no 2: ADME Parameters of standard drug.
|
Standard Erlotinib |
M.F. |
M.W. |
nHBA |
nHBD |
Log (ilogp) |
TPSA |
Rule of Five |
|
Accepted values |
---------------- |
<500 g/mol |
<5 |
<10 |
<5 |
<110 |
Max 4 |
|
1 |
C22H23N3O4 |
393.4 |
6 |
1 |
3.67 |
74.43 |
4 |
Table no 3: ADME Parameters of derived compounds.
|
Compounds |
M.F. |
M.W. |
nHBA |
nHBD |
Log P (ilogp) |
TPSA |
Rule Of Five |
|
Accepted values |
---------------- |
<500 g/mol |
<5 |
<10 |
<5 |
<110 |
Max 4 |
|
1 |
C13H9N3O3 |
255.23 |
4 |
1 |
1.67 |
87.64 |
4 |
|
2 |
C13H14N2O |
214.26 |
2 |
1 |
2.31 |
41.82 |
4 |
|
3 |
C14H9F3N2O |
278.23 |
5 |
1 |
2.3 |
41.82 |
4 |
|
4 |
C15H14N2O3 |
270.28 |
4 |
2 |
2.45 |
71.28 |
4 |
|
5 |
C16H10F3NO4 |
337.25 |
8 |
2 |
2.63 |
75.72 |
3 |
|
6 |
C14H9F3N2O2 |
294.23 |
6 |
1 |
2.61 |
51.05 |
3 |
|
7 |
C14H12N2O |
224.26 |
2 |
1 |
2.29 |
41.82 |
4 |
|
8 |
C14H9N3O |
235.24 |
3 |
1 |
2.01 |
65.61 |
4 |
|
9 |
C14H12N2OS |
256.31 |
2 |
1 |
2.44 |
67.12 |
4 |
|
10 |
C14H12N2O |
224.26 |
2 |
1 |
2.16 |
41.82 |
4 |
STANDARD: (Erlotinib)
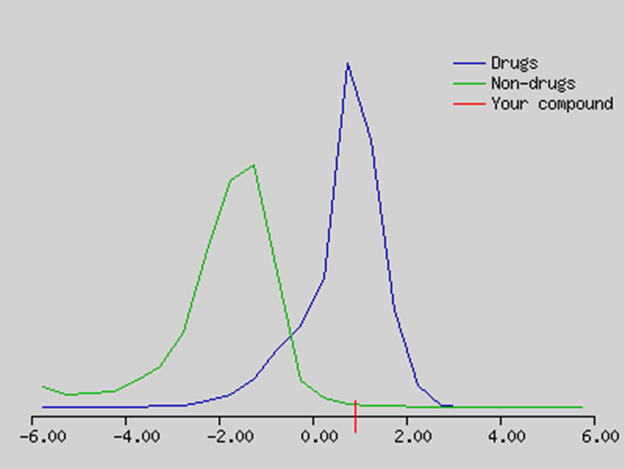
Figure no 3: 2D,3D, structure of erlotinib, drug likeness profile.
Compound
Figure no 4: 2D,3D structures of new molecule.

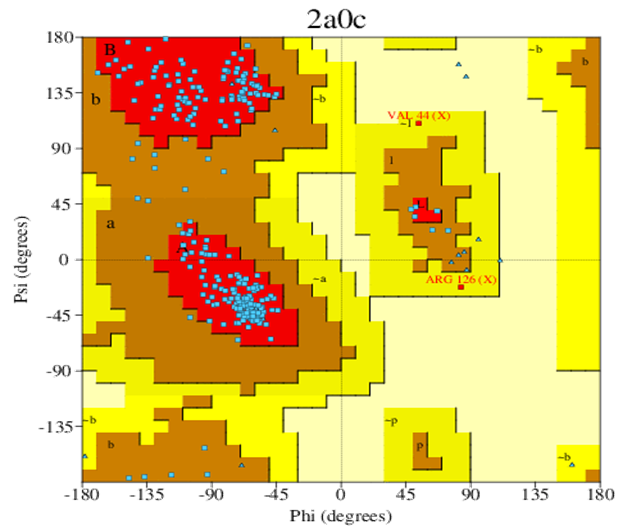
Figure no 5: Protein chain (2a0c), Ramachandran plot.
Table no 4:2D,3D docking results of standard drug.
|
Standard Compound |
2D |
3D |
|
Erlotinib |
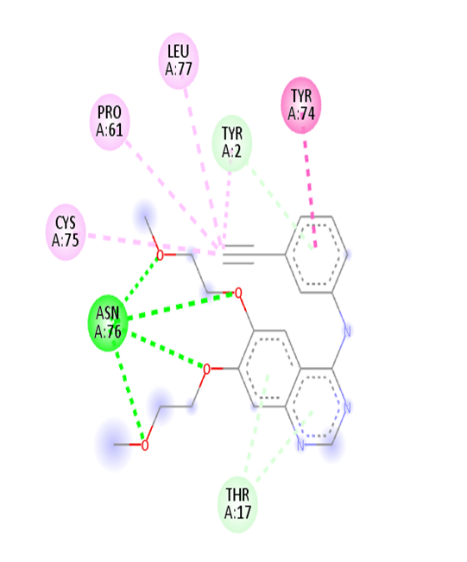
|
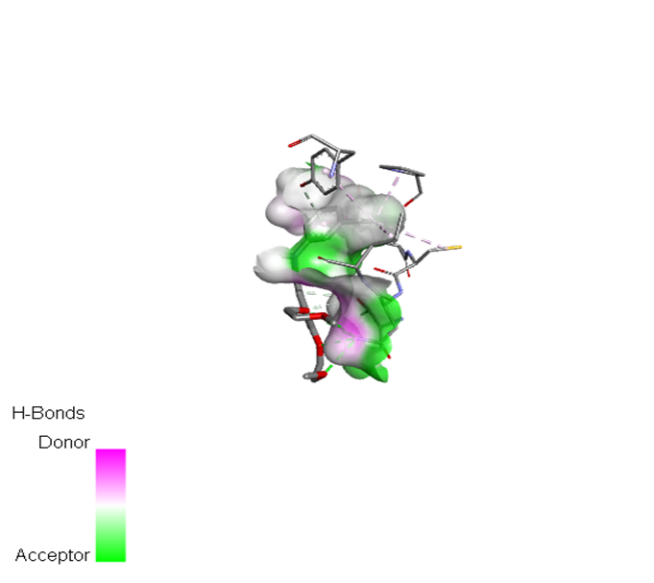
|
Table no 5: 2D,3Ddocking results of derived compounds.
|
Compound |
2D |
3D |
|
1 |
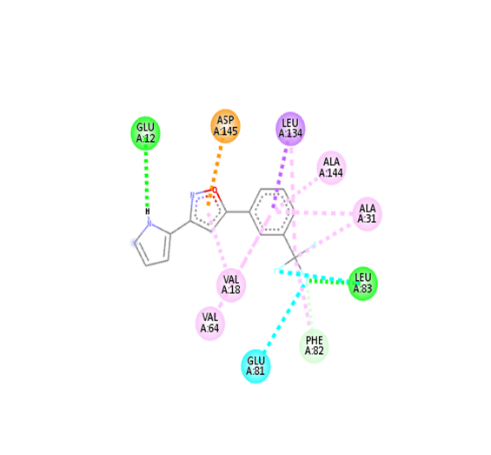
|
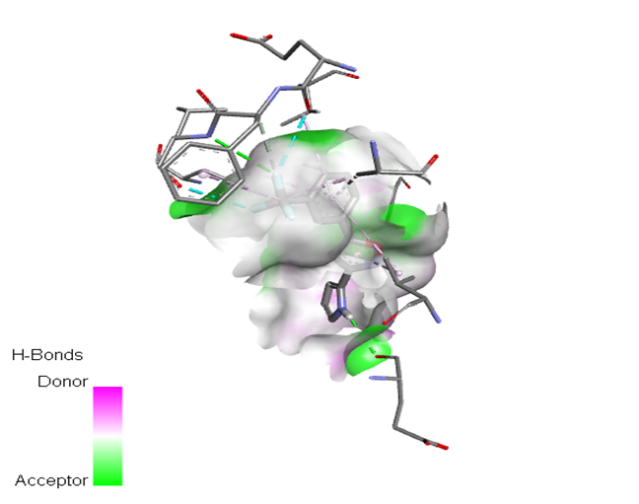
|
|
2 |
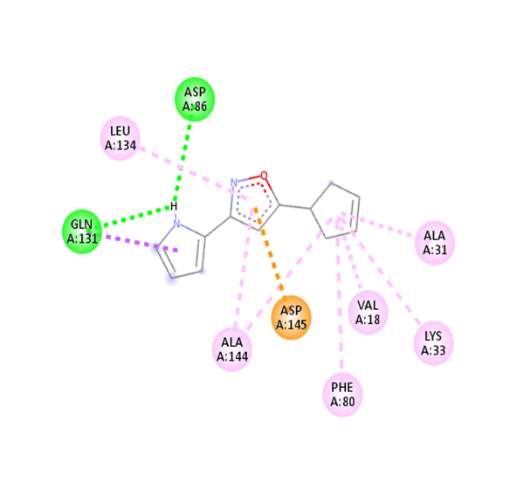
|
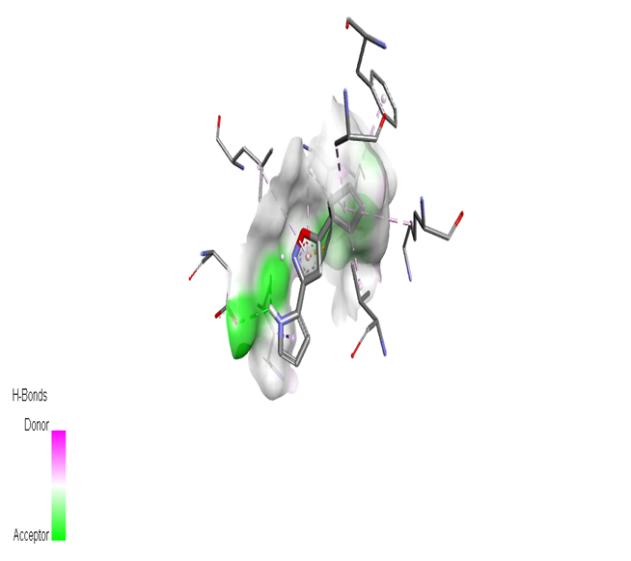
|
|
3 |
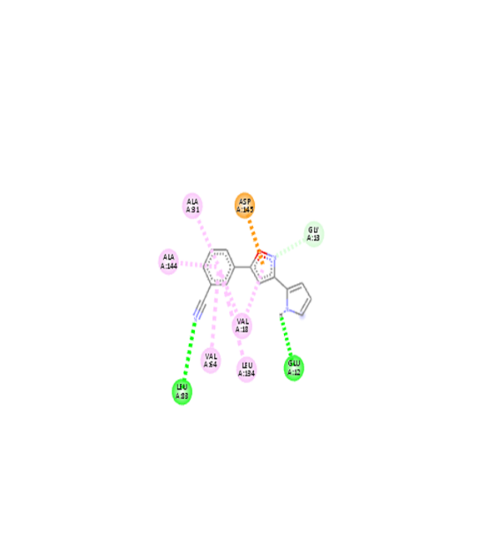
|
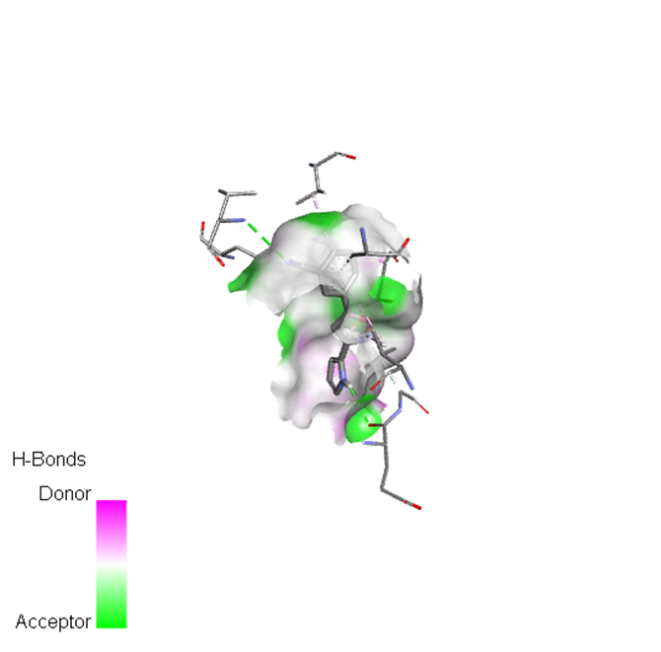
|
|
4 |
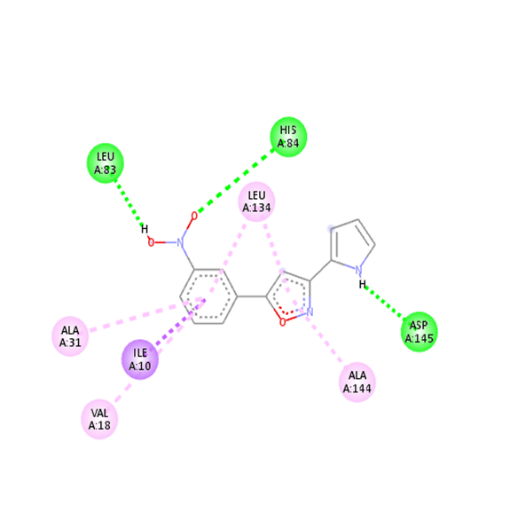
|
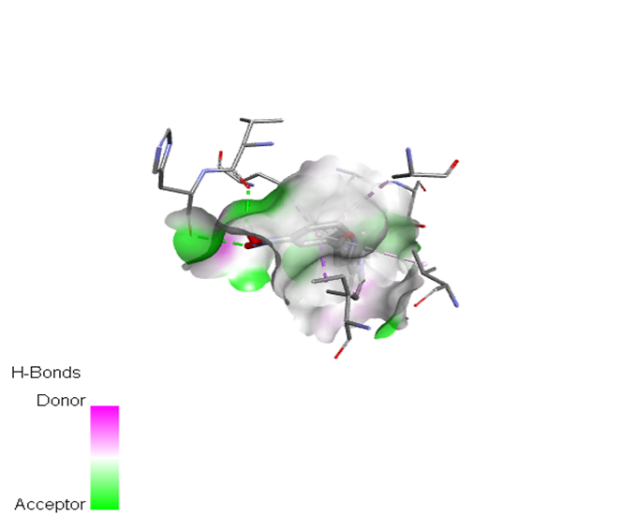
|
|
5 |
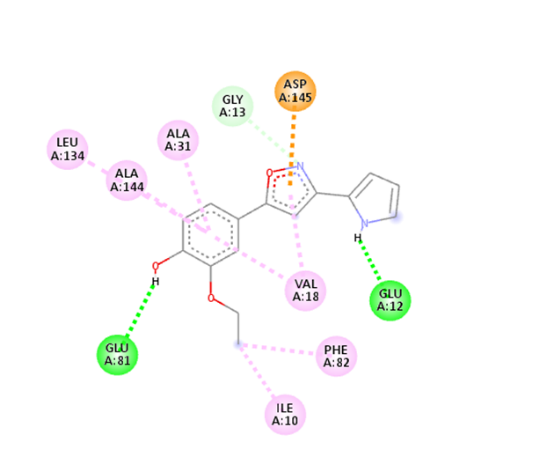
|
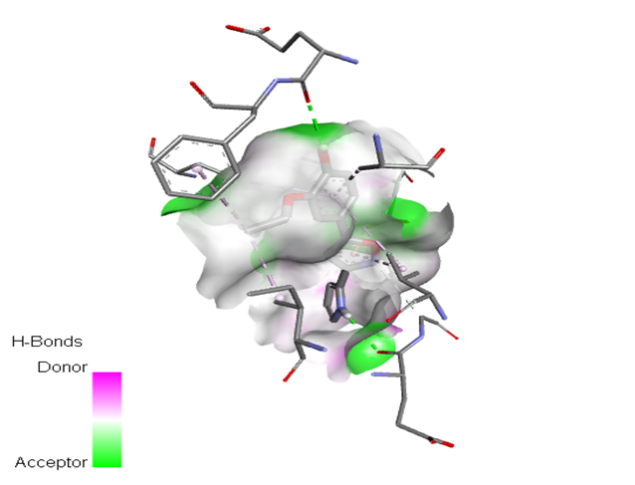
|
|
6 |
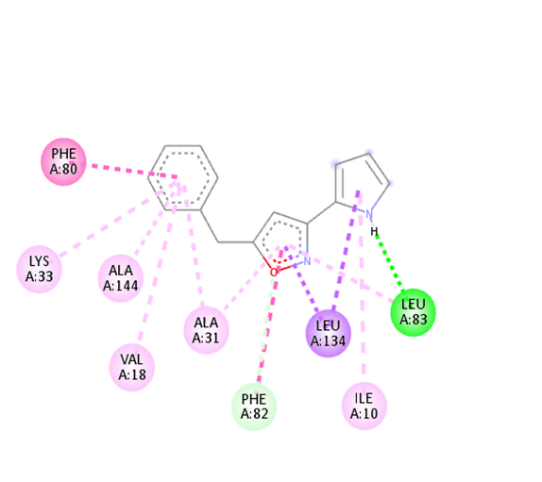
|
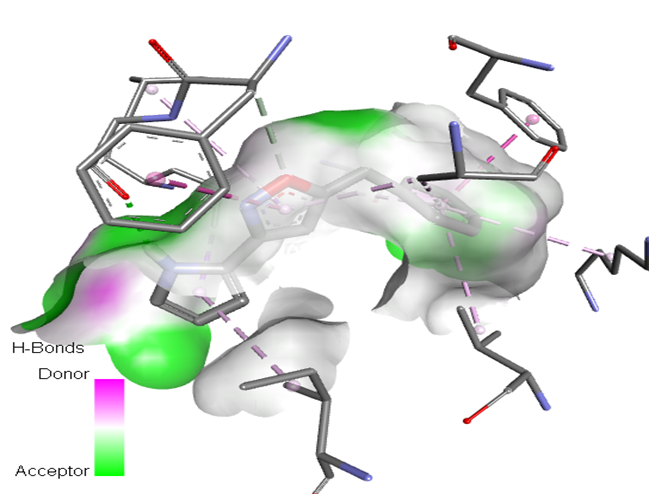
|
|
7 |
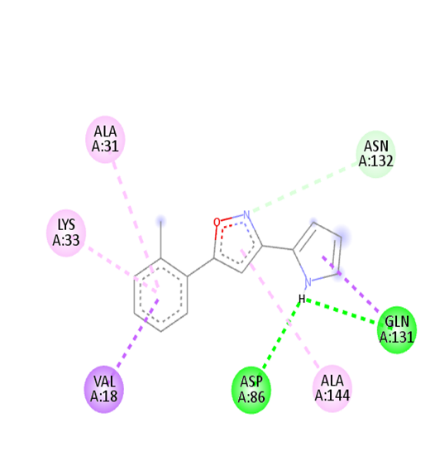
|
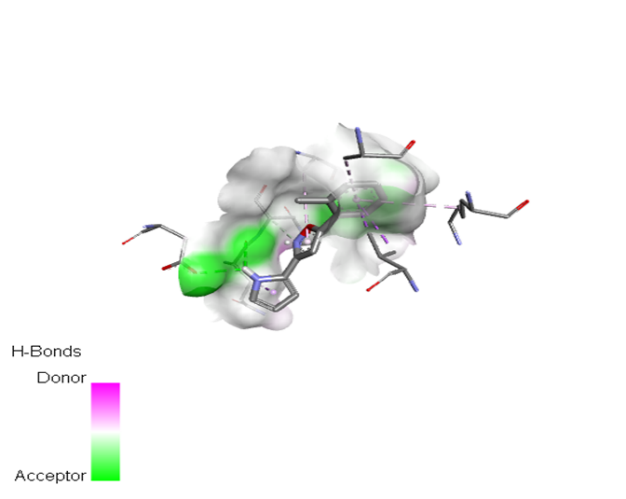
|
|
8 |
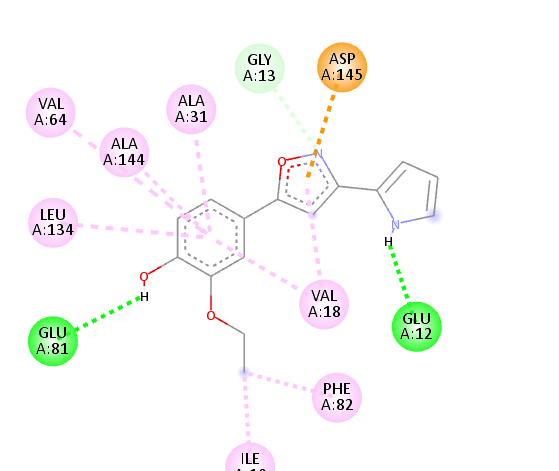
|
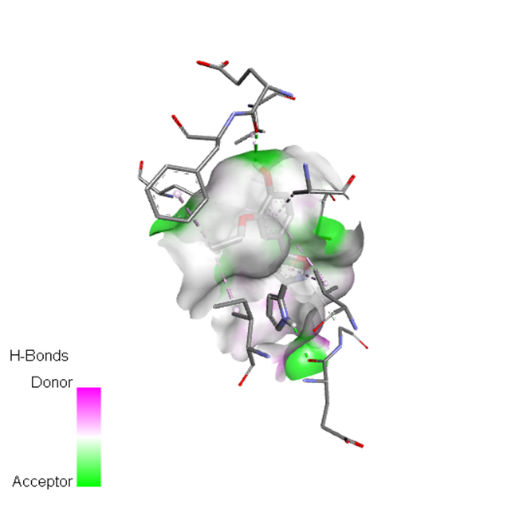
|
|
9 |
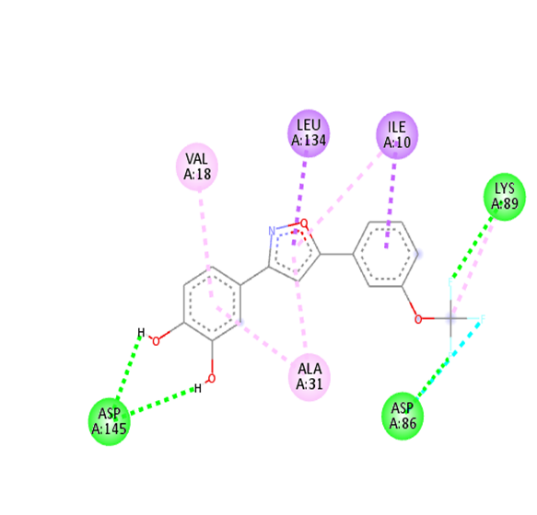
|
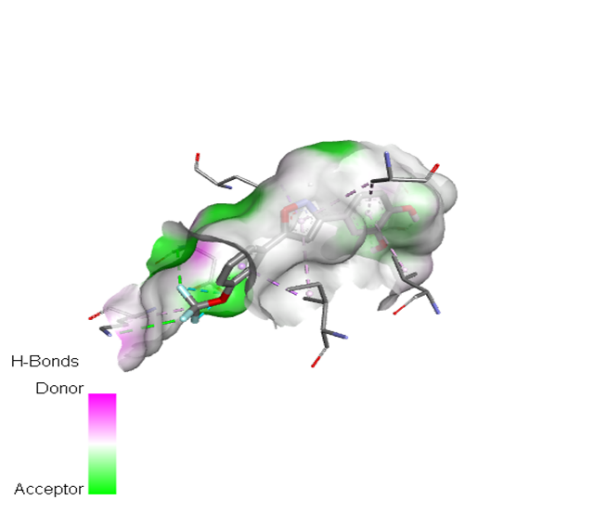
|
|
10 |
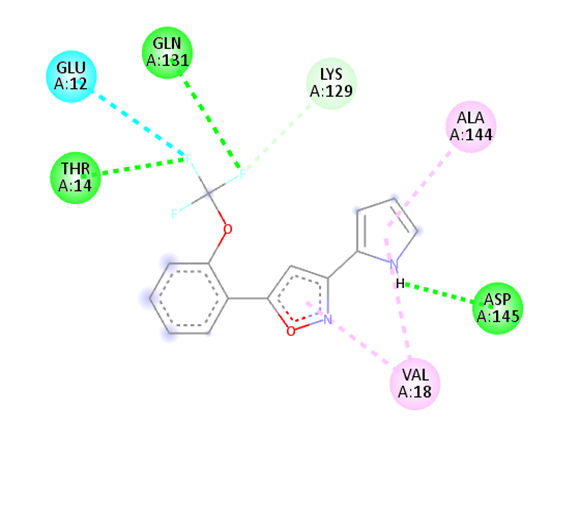
|
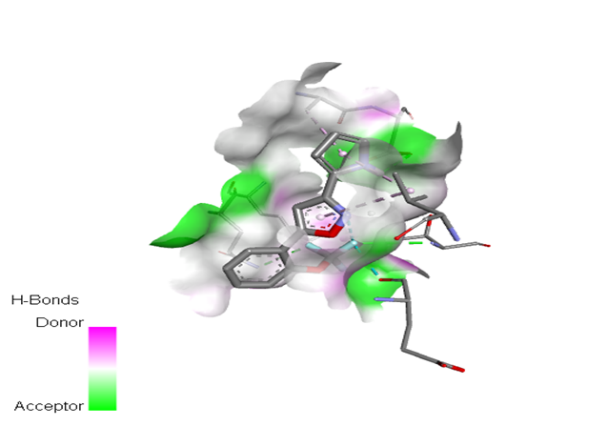
|
Table no 6
|
STANDARD |
PROTEIN |
FREE BINDING ENERGY [Kcal/mol] |
TYPE OF BOND INTERACTED |
INTERACTION GROUP |
LENGTH [ A0 ] |
|
Erlotinib |
1PK4 |
-6.0 |
H-BOND |
A: ASN76 A: ASN76 A: ASN76 A: ASN76 |
3.23246 3.01358 2.82907 2.84363 |
Table no 7
|
COMPOUND |
PROTEIN |
FREE BINDING ENERGY [Kcal/mol] |
TYPE OF BOND INTERACTED |
INTERACTION GROUP |
LENGTH [ A0 ] |
|
1 |
2A0C |
-6.9 |
H bond |
A: LEU83 A: GLU12 |
3.18796 2.61011 |
|
2 |
-6.7 |
A:ASP86 A: GLN131 |
2.67272 2.68438 |
||
|
3 |
-8.4 |
A: LEU83 A: GLU12 |
3.18413 2.62651 |
||
|
4 |
-7.4 |
A: HIS84 A: LEU83 A:ASP145 |
3.27761 2.29344 1.99707 |
||
|
5 |
-7.9 |
A: GLU81 A: GLU12 |
2.3122 2.47253 |
||
|
6 |
-9.1 |
A: LEU83 |
2.14761 |
||
|
7 |
-7.8 |
A:ASP86 A: GLN131 |
2.45995 2.88685 |
||
|
8 |
-8.0 |
A: GLU81 A: GLU12 |
2.40984 2.362 |
||
|
9 |
-8.5 |
A:ASP86 A: LYS89 A:ASP145 A:ASP145 |
3.10521 3.45374 2.0817 2.57026 |
||
|
10 |
-6.4 |
A: THR14 A: GLN131 A:ASP145 A:ASP145 |
3.43755 3.65153 2.71289 2.83705 |
DISCUSSION:
The molecular docking analysis indicated that several of the newly designed compounds exhibited stronger binding affinities than Erlotinib. Compound 6 emerged as the most promising candidate, with the lowest binding energy of –9.1 kcal/mol, forming a hydrogen bond with LEU83, a residue recurrently involved in ligand interactions. This suggests a favorable binding orientation and potentially enhanced inhibitory efficiency. Similarly, Compounds 3 (–8.4 kcal/mol), 9 (–8.5 kcal/mol), and 8 (–8.0 kcal/mol) also demonstrated high binding affinities, forming multiple hydrogen bonds with essential residues such as GLU12, ASP86, and ASP145. These interactions likely enhance the compounds' stability within the ATP-binding pocket, improving their binding specificity of particular interest, Compound 9 formed four hydrogen bonds with ASP86, LYS89, and ASP145, indicating a highly stable interaction network within the active site. This extensive bonding may explain its strong binding affinity and potential bioactivity. Overall, residues including GLU12, LEU83, ASP86, and ASP145 were frequently involved in binding with top-performing compounds, suggesting their critical role in anticancer activity. In contrast, Compound 10 showed the weakest binding among the test compounds, with a binding energy of –6.4 kcal/mol, only slightly better than Erlotinib, indicating limited binding potential.
CONCLUSION:
Novel isoxazole derivatives were subjected to molecular docking investigations and in silico ADMET prediction, which demonstrated significant binding affinity for the target protein and promising drug-like characteristics. Good absorption, low toxicity, and sustained interactions within the binding site were among the drugs' positive pharmacokinetic characteristics. These results imply that the isoxazole derivatives under study have the potential to serve as lead molecules for additional medication development. To confirm their pharmacological efficacy and safety, more experimental research is necessary. To move these molecules closer to therapeutic uses, it will be crucial to combine computational and experimental methods. All the molecules demonstrate favourable results relative to the standard drug. The preceding information indicates that all drugs have affinity for the protease receptor, which is crucial in the pathophysiology of cancer. As a result, we can proceed with IN-VIVO and IN-VITRO investigations.
REFERENCES


Arya Kate, Pranav Sargar, Viganesh Manugade, Shreya Gouraje, Rohan Patil, Molecular Docking and ADMET Profiling of Novel Isoxazole-Based Compounds for Cancer Therapy, Int. J. of Pharm. Sci., 2025, Vol 3, Issue 9, 2990-3001. https://doi.org/10.5281/zenodo.17200936
 10.5281/zenodo.17200936
10.5281/zenodo.17200936