Department of Pharmaceutical Chemistry, Bhupal Nobles University, Udaipur, Rajasthan.
For decades, the pharmaceutical industry has relied on occupancy-driven pharmacology, limiting drug discovery to proteins with accessible binding pockets, representing only ~20% of the human proteome. Targeted Protein Degradation (TPD) has emerged as a transformative paradigm, shifting the focus from inhibition to elimination. This review critically analyzes the evolution from first-generation Proteolysis Targeting Chimeras (PROTACs) to next-generation degraders and molecular glues. We discuss how novel E3 ligase recruitment, expanding beyond cereblon and VHL, is enhancing tissue selectivity and overcoming resistance mechanisms. Furthermore, we examine the paradigm shift from "accidental" discovery to rational design of molecular glues, which offer improved pharmacokinetic properties compared to bivalent degraders. By highlighting recent breakthroughs in targeting transcription factors, scaffold proteins, and extracellular domains, we illustrate how TPD is successfully expanding the druggable proteome, offering new therapeutic avenues for previously intractable diseases
The "druggable genome" concept has historically constrained drug discovery to proteins possessing distinct, high-affinity binding pockets, such as G-protein-coupled receptors (GPCRs) and kinases. Conventional small molecules act via an occupancy-driven mechanism, requiring high systemic concentrations to maintain therapeutic efficacy, often leading to off-target toxicity and the development of resistance. Approximately 80% of the human proteome, including transcription factors and scaffold proteins, remains "undruggable" due to the lack of deep enzymatic pockets[1].
Targeted Protein Degradation (TPD) represents a paradigm shift from inhibition to elimination. By harnessing the body's natural disposal system—the ubiquitin-proteasome system (UPS)—TPD modalities induce proximity between an E3 ubiquitin ligase and a target protein, leading to polyubiquitination and subsequent proteasomal degradation. This "event-driven" pharmacology offers distinct advantages: it targets protein function rather than activity, can degrade overexpressed or mutant proteins, and operates catalytically, allowing for sub-stoichiometric dosing [2].
While first-generation degraders focused primarily on well-characterized targets and E3 ligases (Cereblon and VHL), the field is now entering a new era. This review focuses on "next-generation" technologies: the expansion of the E3 ligase toolbox, the rational design of molecular glues, and the exploration of non-proteasomal degradation pathways, all of which are pushing the boundaries of the druggable proteome.
2. Next-Generation PROTACs: Beyond CRBN and VHL
Early PROTACs relied heavily on the E3 ligases Cereblon (CRBN) and Von Hippel-Lindau (VHL). While successful, this reliance presents limitations. First, CRBN and VHL are ubiquitously expressed, potentially causing systemic degradation and toxicity. Second, some cancers downregulate these ligases to develop resistance. Next-generation strategies are addressing these bottlenecks.
2.1 Expanding the E3 Ligase Toolbox
The human genome encodes over 600 E3 ligases. Next-generation PROTACs are now recruiting tissue-specific E3 ligases to improve the therapeutic index.
Tissue-Specific Degraders: Recent studies have utilized E3 ligases such as RNF4 and DCAF15, which show differential expression profiles across tissues. For instance, DCAF15 is the mechanistic target of the sulfonamide drugs (e.g., indisulam), offering a pathway for degradation in specific tumor subtypes [3].
Molecular "SWITCH" Technology: Researchers are developing ligands for E3 ligases like MDM2 and IAP, which possess distinct cellular localizations and substrate specificities, allowing for the degradation of targets previously resistant to CRBN/VHL-based PROTACs[4].
2.2 Overcoming Resistance
Resistance to PROTACs can arise via genomic alterations in the E3 ligase complex components. Next-generation degraders utilize "E3-agostic" approaches, where the same warhead can be conjugated to different E3-recruiting ligands. This modular capability allows for the rapid generation of backup compounds should resistance to a specific E3 ligase emerge[5]
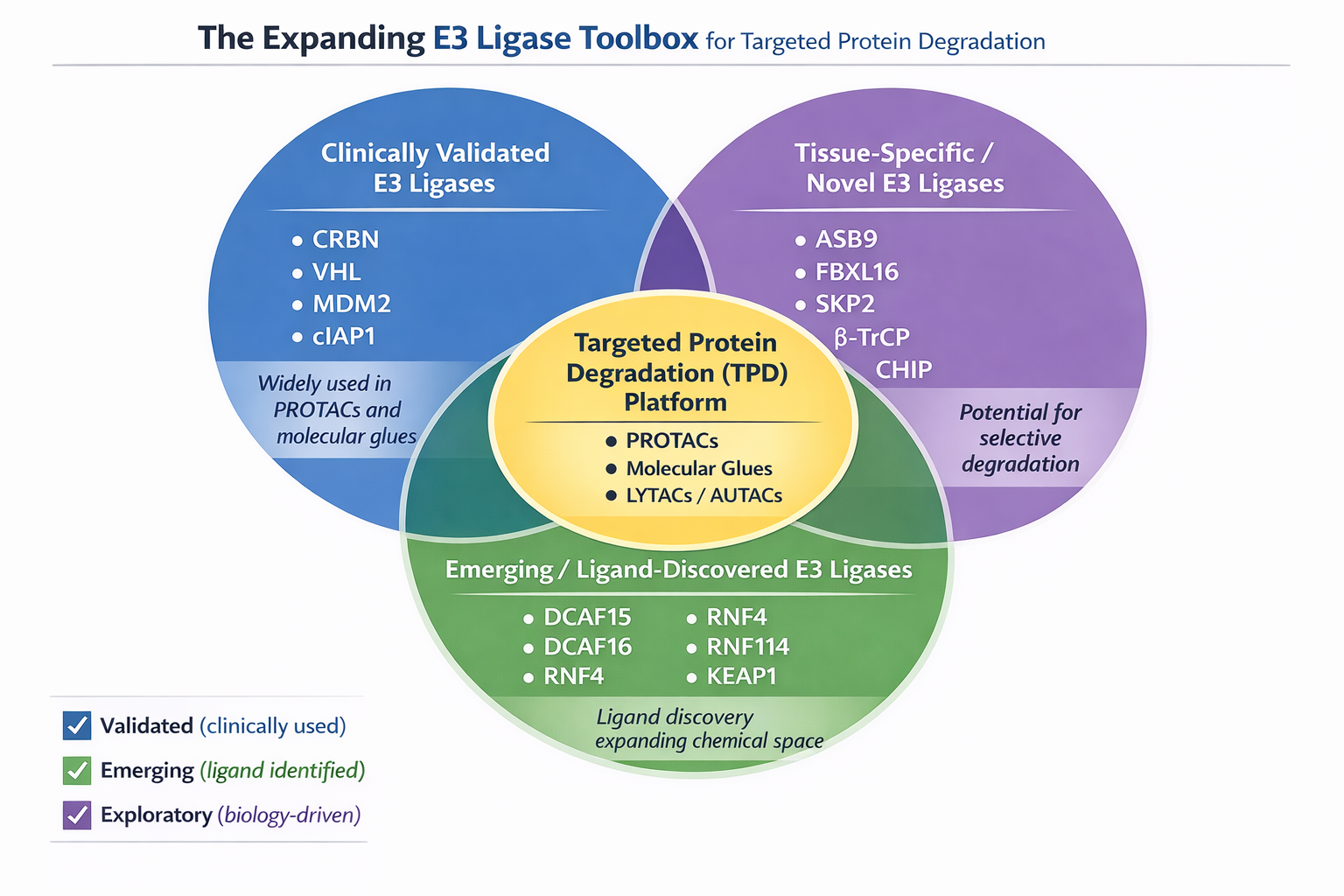
Figure 1. The Expanding E3 Ligase for Targeted Protein Degradation
3. Molecular Glues: The "Bivalent" Alternative
While PROTACs are typically large, bivalent molecules with molecular weights exceeding 1000 Da, molecular glues are smaller, monovalent compounds that stabilize protein-protein interactions (PPIs). They represent a more drug-like class of degraders with higher potential for oral bioavailability and blood-brain barrier penetration.
3.1 From Serendipity to Rational Design
Historically, molecular glues were discovered serendipitously; thalidomide and its analogs were found to act as glues only decades after their introduction. However, the field is transitioning toward rational design.
Structure-Based Discovery: Recent cryo-EM and X-ray crystallography studies have elucidated the structural motifs required for "glue" activity. This has led to the identification of non-thalidomide scaffolds capable of inducing novel CRBN-neosubstrate interactions [6].
Phenotypic Screening: High-throughput screening campaigns specifically designed to identify degrader phenotypes are uncovering glues for targets previously thought undruggable, such as the transcription factor CDK8 [7].
3.2 Cyclin-Dependent Kinases (CDKs) and Beyond
A landmark achievement in the field was the discovery of glues targeting Cyclin K. Unlike traditional inhibitors, these compounds induce the degradation of Cyclin K by recruiting the DDB1-CUL4-RBX1 E3 ligase complex, independent of CRBN. This highlights the potential of glues to rewire cellular machinery for therapeutic benefit [8].
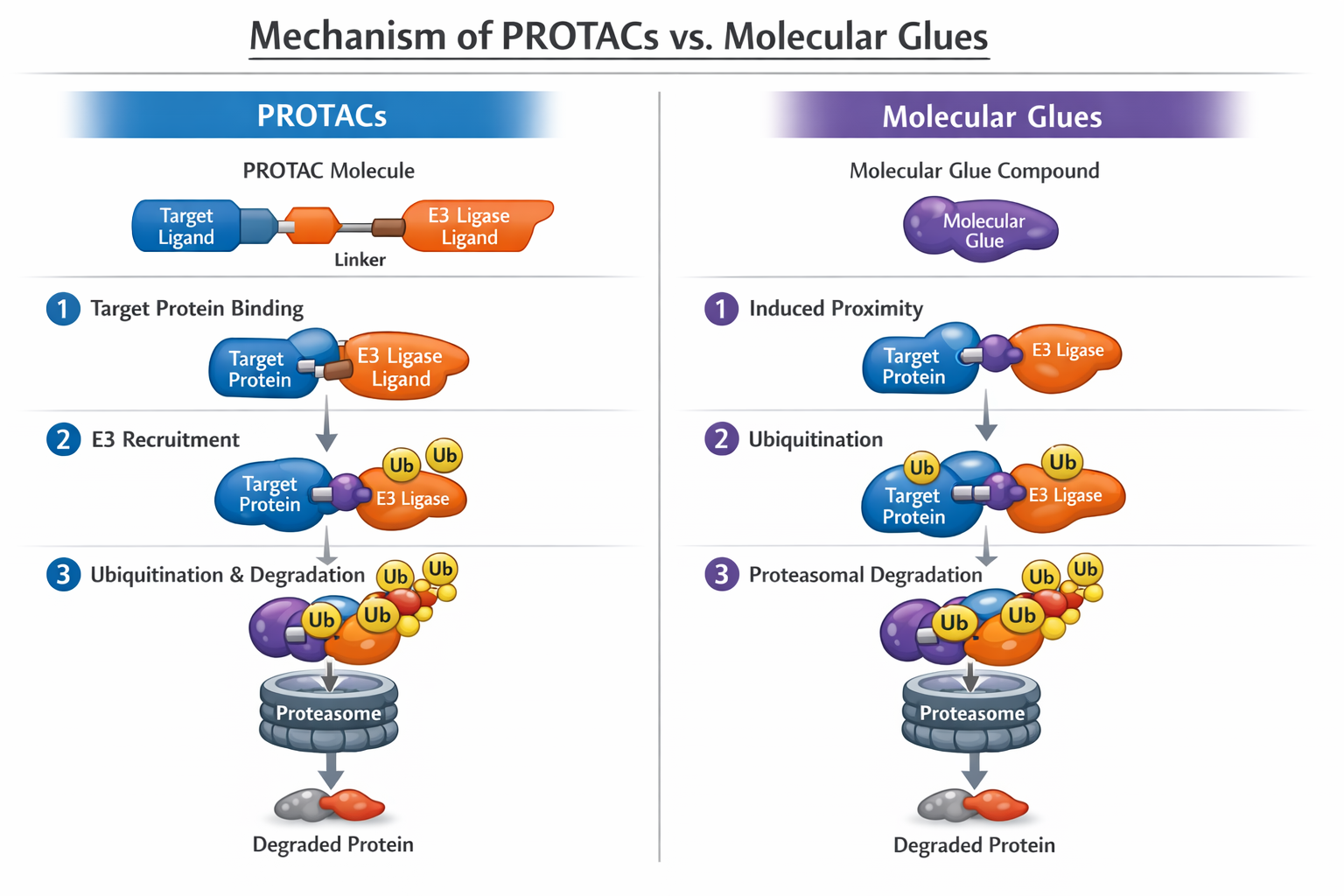
Figure 2. Mechanism of PROTACs vs. Molecular Glues
4. Expanding the Druggable Proteome
The ultimate metric of success for TPD is its ability to target the "undruggable." Next-generation degraders are making significant inroads into three critical classes of proteins.
4.1 Transcription Factors
Transcription factors (TFs) lack enzymatic pockets and possess large protein-protein interaction surfaces, making them recalcitrant to small-molecule inhibition. PROTACs have successfully degraded TFs such as STAT3, c-Myc, and the androgen receptor (AR). By targeting the protein for destruction rather than inhibiting its activity, PROTACs bypass the need for high-affinity binding to the DNA-binding domain, often requiring only weak ligands to initiate the degradation cascade [9].
4.2 Scaffold Proteins
Scaffold proteins, which facilitate signaling complexes but lack catalytic activity, are another frontier. For example, the degradation of the scaffold protein FAK (Focal Adhesion Kinase) via PROTACs has shown efficacy in reducing tumor metastasis, distinct from the effects of FAK kinase inhibitors [10].
4.3 Extracellular and Membrane Proteins
While the UPS is intracellular, next-generation technologies are extending degradation to the cell surface.
LYTACs (Lysosome-Targeting Chimeras): These bifunctional molecules bind extracellular proteins (e.g., immune checkpoints like PD-L1) and engage the lysosomal targeting receptor (CI-M6PR), routing the protein to the lysosome for degradation. This expands the TPD modality to secreted factors and membrane receptors [11].
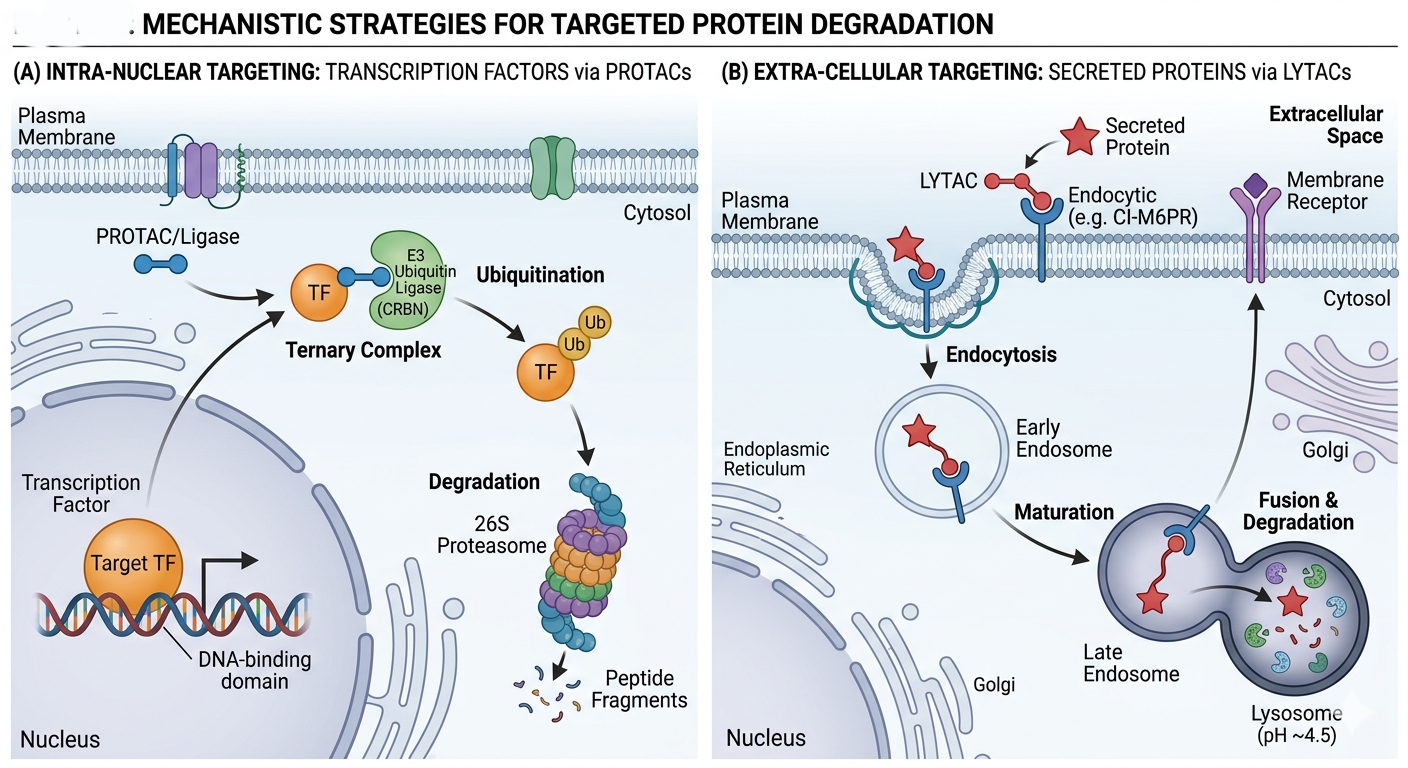
Figure 3. Mechanistic Strategies for Targeted Protein Degradation
5. Challenges and Future Perspectives
Despite rapid progress, significant hurdles remain for the clinical translation of next-generation degraders.
5.1 Pharmacokinetics and Drug Delivery
PROTACs typically violate Lipinski's "Rule of 5," leading to poor solubility and permeability. Future research must focus on formulation strategies, prodrug approaches, and alternative routes of administration (e.g., intrathecal or inhaled) to maximize tissue exposure [12].
5.2 Toxicity and the "Hook Effect"
The "Hook effect"—where high concentrations of a PROTAC saturate the binary interactions (Target-PROTAC or PROTAC-E3) without forming the productive ternary complex—remains a pharmacodynamic challenge. Furthermore, off-target degradation (neo-substrate recruitment) remains a safety concern, necessitating rigorous proteomic screening of candidate molecules[13].
5.3 The Future: Non-Proteasomal Degradation
The field is looking beyond the proteasome. Emerging modalities like AUTACs(Autophagy-Targeting Chimeras) and ATTECs target the autophagy machinery, enabling the degradation of larger aggregates, organelles, and intracellular pathogens, further expanding the scope of degradable targets [14].
CONCLUSION
Next-generation PROTACs and molecular glues are revolutionizing our approach to drug discovery. By moving beyond the limitations of the "druggable pocket" and expanding the repertoire of E3 ligases, TPD is unlocking the therapeutic potential of the "undruggable" proteome. The transition from serendipitous discovery to rational design, coupled with the exploration of non-proteasomal pathways, promises to deliver a new wave of medicines for cancers, neurodegenerative diseases, and immune disorders. As these technologies mature, they will likely become a cornerstone of modern medicinal chemistry.
REFERENCES



Shrey Parmar, Dr. Yuvraj Singh Sarangdevot, Sadaf Ashraf, Next-Generation PROTACs and Molecular Glues: Expanding the Druggable Proteome, Int. J. of Pharm. Sci., 2026, Vol 4, Issue 4, 393-397 https://doi.org/10.5281/zenodo.19395854
 10.5281/zenodo.19395854
10.5281/zenodo.19395854
