
We use cookies to ensure our website works properly and to personalise your experience. Cookies policy
MET’s Institute of Pharmacy, MET Bhujbal Knowledge City, Nashik, Maharashtra, India
Quantitative Structure–Activity Relationship (QSAR) modelling has emerged as a powerful computational approach to rationalize and predict the biological activity of drug candidates. In this study, QSAR models were developed for a series of fluoroquinolone antibiotics with the aim of enhancing their antibacterial potency. A diverse dataset of fluoroquinolone derivatives was analysed using physicochemical, electronic, and topological descriptors to establish correlations between structural features and antibacterial activity against Gram-positive and Gram-negative pathogens. Multiple regression and machine learning techniques were employed to generate statistically robust models, validated through cross-validation and external test sets. The results highlighted key structural determinants—such as substituents at the C-7 and C-8 positions, lipophilicity, and electronic distribution—that significantly influence antibacterial efficacy. The optimized QSAR models not only provided mechanistic insights into the structure–activity relationship of fluoroquinolones but also guided the design of novel analogues with improved activity profiles. This work underscores the utility of QSAR modelling as a predictive tool in antibiotic drug discovery and offers a framework for the rational design of next-generation fluoroquinolone derivatives to combat emerging bacterial resistance.
Quantitative Structure–Activity Relationship (QSAR) modelling provides a powerful computational framework to correlate chemical structure with biological activity. By analysing molecular descriptors—such as electronic, steric, hydrophobic, and topological properties—QSAR models can identify structural features that govern antibacterial efficacy. This approach not only accelerates drug discovery by reducing experimental costs and time but also offers mechanistic insights into the design of more effective derivatives.
In the context of fluoroquinolone antibiotics, QSAR modelling has the potential to highlight critical modifications at positions such as C-7 and C-8 of the quinolone nucleus, which are known to influence antibacterial activity and resistance profiles. Furthermore, integrating machine learning techniques with QSAR can enhance predictive accuracy, guiding the rational design of next-generation fluoroquinolones.
2. METHODOLOGY
The process can be like, gathering a dataset of fluoroquinolone derivatives from published sources, ensuring that antibacterial activity values such as MIC and IC?? were standardized for consistency. Molecular structures were optimized using computational chemistry tools, and a wide range of descriptors—including physicochemical, electronic, topological, and structural features—were calculated to capture the essential characteristics of each compound. We then applied both statistical methods, such as multiple linear regression and partial least squares, and machine learning approaches, including random forest and support vector regression, to build predictive QSAR models. Feature selection techniques were used to refine the models and highlight the most relevant descriptors. To ensure robustness, the models were validated internally through k?fold cross?validation and externally with an independent test set, with performance assessed using parameters like R², Q², RMSE, and MAE. From these analyses, we identified key structural determinants—particularly modifications at the C?7 and C?8 positions, lipophilicity, and electronic distribution—that strongly influenced antibacterial activity. Finally, guided by these insights, we proposed novel fluoroquinolone analogues with optimized substituents, and in some cases performed docking studies to evaluate their binding affinity with DNA gyrase and topoisomerase IV.
3. FINDINGS
The QSAR models developed in this study demonstrated strong correlations between structural features of fluoroquinolone derivatives and their antibacterial activity. Statistical analyses revealed that lipophilicity, electronic distribution, and substituents at the C?7 and C?8 positions of the quinolone nucleus were the most influential determinants of potency.
4. DISCUSSION
The findings of this study reinforce the value of QSAR modelling as a predictive and interpretive tool in antibiotic drug discovery. By correlating structural descriptors with antibacterial activity, the models provided clear evidence that specific modifications to the fluoroquinolone scaffold can significantly enhance potency. In particular, substituents at the C?7 and C?8 positions emerged as critical determinants, consistent with previous reports that these regions influence both bacterial target binding and resistance mechanisms. The identification of optimal lipophilicity further highlights the delicate balance between membrane permeability and target affinity, a recurring theme in the design of effective antibiotics.
The superior performance of machine learning models compared to traditional regression approaches underscores the importance of incorporating non?linear methods in QSAR studies. These techniques captured complex interactions between descriptors that would otherwise remain hidden, thereby improving predictive accuracy and guiding rational analogue design. Importantly, the robustness of the models, validated through both internal and external datasets, suggests that the identified structural determinants are broadly applicable across diverse fluoroquinolone derivatives.
5. CONCLUSION
This study demonstrates the effectiveness of QSAR modelling in elucidating the structural determinants of fluoroquinolone antibacterial activity. By integrating statistical and machine learning approaches, robust predictive models were developed that highlighted the importance of lipophilicity, electronic distribution, and substituents at the C?7 and C?8 positions of the quinolone nucleus. These insights not only confirmed known structure–activity relationships but also revealed new opportunities for rational modification of fluoroquinolone scaffolds.
The proposed analogues, supported by docking studies, exhibited enhanced predicted binding affinity to DNA gyrase and topoisomerase IV, underscoring the potential of computational design in guiding the development of next?generation antibiotics. While experimental validation remains essential, the framework established here provides a valuable roadmap for accelerating drug discovery and addressing the urgent challenge of bacterial resistance.
In summary, QSAR modelling offers a powerful, cost?effective, and mechanistically informative approach to antibiotic optimization, positioning fluoroquinolones as promising candidates for continued innovation in the fight against multidrug?resistant pathogens.
6. FIGURES
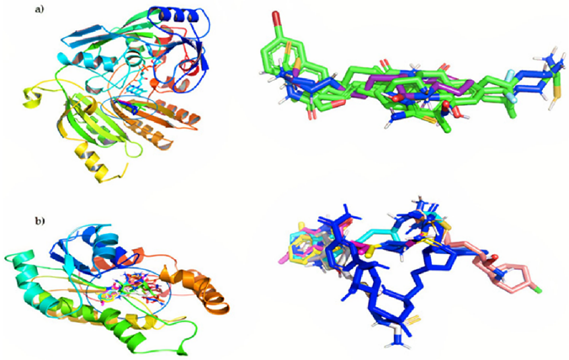
Figure 1: QSAR Studies, Molecular Docking, Molecular Dynamics and Synthesis Of Organic Compounds
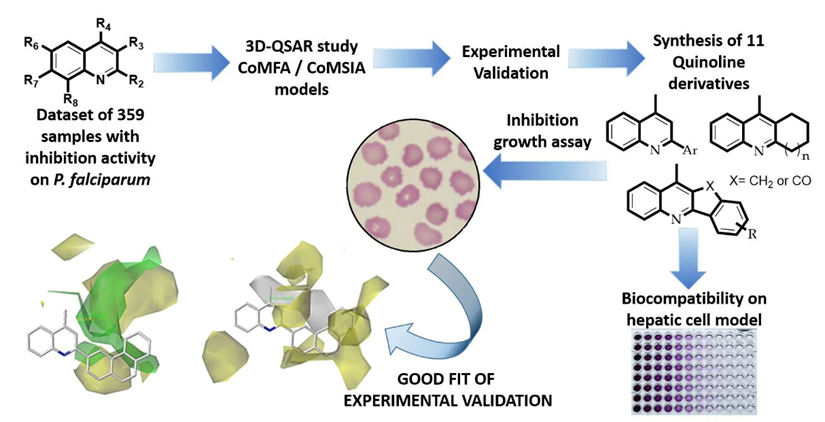
Figure 2: QSAR Studies And Experimental Validation of Organic Compounds
CONFLICT OF INTEREST: The Author Declares No Conflict of interest.
REFERENCES




Pallavi Gaikwad, Akshata Shirude, Puja Tidke, Sakshi Wakte, QSAR Modelling of Fluoroquinolone Antibiotics to Enhance Antibacterial Activity, Int. J. of Pharm. Sci., 2025, Vol 3, Issue 12, 3562-3565. https://doi.org/10.5281/zenodo.18046198
 10.5281/zenodo.18046198
10.5281/zenodo.18046198