
We use cookies to ensure our website works properly and to personalise your experience. Cookies policy
Lokmanya Tilak Institute of Pharmacy, Navi Mumbai, Maharashtra, India 410210
From conventional treatments to sophisticated molecular and computational approaches, drug research and development is a difficult, expensive, and time-consuming process. Natural products and serendipity have historically been the foundation of discoveries, which have since developed through synthetic chemistry, molecular biology, and biotechnology. Drug resistance, high attrition rates, safety concerns, and rising development costs are still major obstacles in spite of these advancements. Every step of the drug development process can benefit from the quicker and more effective solutions that artificial intelligence (AI) offers. AI improves the design of clinical trials and the discovery of biomarkers while enabling quick target identification, generative molecular design, virtual screening, and predictive modeling of pharmacokinetics and toxicity.Rentosertib, the first AI-designed medication to show clinical proof-of-concept, is a significant milestone that highlights how AI can shorten development times from decades to a few years. The historical context, traditional approaches, and contemporary AI applications in drug discovery are examined in this review, along with their benefits, drawbacks, and potential future developments. The pharmaceutical sector may create more accurate, cost-effective, and customized treatments by carefully integrating AI with conventional knowledge, transforming healthcare around the world.
A drug development program begins when there is a disease or clinical condition for which no acceptable medicinal products exist, and this unmet clinical need serves as the project's driving reason. The first study, which is frequently conducted in academia, provides data to form a hypothesis that inhibiting or activating a protein or pathway may result in a therapeutic impact in a disease state. This activity results in the identification of a target, which may require additional validation before progressing to the lead discovery phase to support a drug development effort. During lead discovery, an aggressive search is conducted to locate a drug-like small molecule or biological treatment, known as a development candidate, that will go into preclinical, and if successful, clinical development and eventually become a marketable pharmaceutical [1]. The timeline below summarizes the major events in drug discovery and development, from early herbal practices to modern biotechnology advancements (Fig. 1)
History
1. Drug development in prehistoric times.
Plants have been utilized as remedies since the Paleolithic era [2]. The Ebers Papyrus (1536 BCE) from Egypt documented hundreds of plant treatments, while Greek physicians such as Hippocrates and Disocorides lay the groundwork for rational medicine and pharmacology [3-6]. Between the 9th and 13th centuries, Arabic intellectuals improved this knowledge by establishing apothecaries and defining the pharmacist's duty [7]. In India, Ayurveda recorded around 700 therapeutic plants in Charak and Sushrut Samhita, while in China, Traditional Chinese Medicine emerged from the Han Dynasty with the Compendium of Materia Medica, which now includes over 11,000 herbs [8-9].
2. Drug discoveries during the 18th and 19th centuries.
James Lind's controlled clinical trial on scurvy in 1753 started the idea of evaluating how well medicines work. By the early 1800s, strong chemicals like morphine, quinine, and atropine had been extracted from plants. Morphine was the best pain reliever for severe pain. Soon after, progress in synthetic chemistry led to the creation of aspirin from salicylic acid, which became one of the most commonly used medications worldwide. [6, 11]
3. Drug discovery in 20th century
In the 20th century, the production of drugs not derived from plants began to emerge. During the early 1900s, doctors used heparin, which was extracted from dog livers, to treat blood clots. They used insulin from dog pancreases for severe diabetes. Antibiotic medications like penicillin, ciclosporin, and tacrolimus came from fungi, while streptomycin and tetracyclines were sourced from soil bacteria. Discovered by Alexander Fleming in 1928, penicillin was the first antibiotic and has saved countless lives [12]. In 1906, the first successful direct blood transfusion between humans during surgery was performed, marking the beginning of cell therapy. In 1939, the inaugural bone marrow transplantation aimed at treating aplastic anaemia was carried out, paving the way for research in bone transplantation therapy. After several decades of research, the first bone transplantation between an unrelated donor and recipient represented another milestone in cell therapy. In 1981, murine embryos were utilized to create the first stem cell, and the potential of stem cells in cell therapy has been extensively examined ever since [14].
4. Late 20s
By the late twentieth century, developments in molecular biology and recombinant DNA technology had revolutionized drug research, allowing for the synthesis of recombinant proteins like human insulin ("Humulin," 1982), which were safer and more scalable than natural proteins. Hybridoma technology (1975) accelerated the development of monoclonal antibodies, and by 2000, antibodies accounted for approximately 25% of novel pharmaceuticals. The Human Genome Project enhanced target-based medication design, while biopharmaceuticals grew to encompass growth hormones, blood factors, and thrombolytics, with more than 120 authorized by 2002. Biogenerics first appeared in 2006 with Sandoz's Omnitrope, and recombinant insulin, erythropoietin, and interferons quickly followed. Stem cell technology, which began with the discovery of human embryonic stem cells in 1998, have expanded the potential for predicting medication toxicity, lowering reliance on animal testing, and developing regenerative medicine. Overall, modern drug research today combines molecular biology, genetics, chemistry, and biotechnology to create, test, and provide safer and more effective medicines. [15]
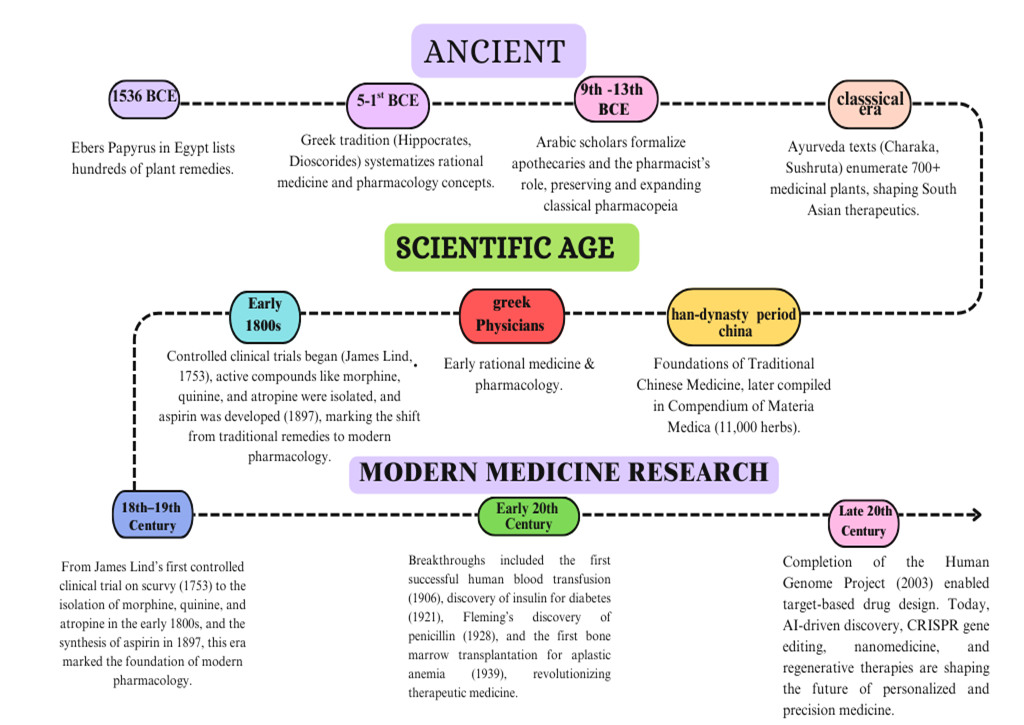
Figure 1: Timeline of Major Milestones in Drug Discovery and Development.
Strategic Approaches to Improve Drug Discovery and Development
The pharmaceutical industry has created comprehensive strategic methods to increase success rates and efficiency in drug research and development, tackling important obstacles such as high prices, long delays, and significant failure rates. Seven major methods have emerged as basic cornerstones of improved R&D productivity (Fig.2): genomics and proteomics. Exploitation uses molecular understanding of illnesses to select and evaluate therapeutic targets more precisely [16, 17]. Proteomics, the substrate of genomics, has been this pathway of drug development has been extensively studied. Proteomics identifies, characterizes, and quantifies cellular proteins to understand their function in disease development and potential for chemotherapeutic intervention [18, 19]. Comparative phenotypic and target-based screening platforms combine empirical whole-cell approaches that excel at identifying first-in-class molecules with hypothesis-driven target-based methods that create superior follower compounds to establish a synergistic discovery framework [20]. Drug repurposing and repositioning techniques speed up development by discovering new therapeutic uses for existing molecules, lowering development time and costs while salvaging previously abandoned compounds [21]. Precompetitive partnerships, pharma-academia alliances, and public-private collaborations are examples of collaborative research strategies that maximize resource utilization and allow corporations to focus on core capabilities while gaining access to specialized skills [22]. Strategic focus on underserved therapeutic areas, such as neglected tropical diseases and rare conditions, presents opportunities for novel target discovery while benefiting from regulatory incentives and reduced competition [23]. Outsourcing strategies combined with pharmaceutical modelling and artificial intelligence enable companies to access specialized capabilities through contract research organizations while leveraging computational approaches, virtual screening These integrated techniques constitute a paradigm change toward data-driven, collaborative, and technologically advanced drug development, addressing both scientific complexity and economic sustainability in current pharmaceutical R&D [22].
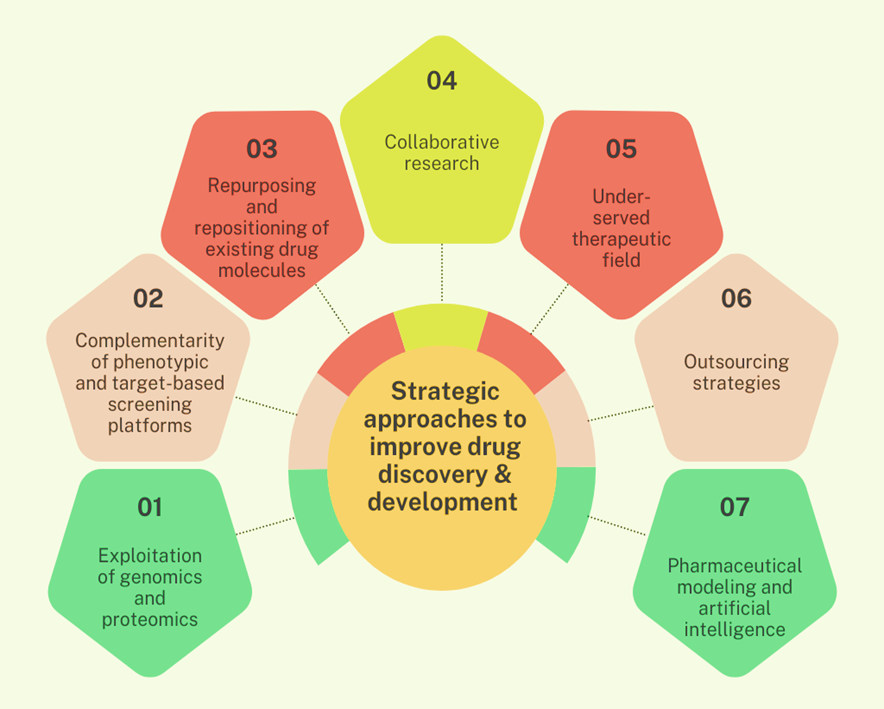
Figure 2: Strategic Approaches to Enhance The Drug Discovery and Development Process
Challenges and complexity of drug development
Drug design involves several hurdles, including commercial viability, clinical translation, and molecular complexity. Most new medications target narrow patient groups, such as cancer and orphan illnesses, limiting blockbuster potential while competing against generics for established uses. Phase II failures are mostly caused by a lack of effectiveness and safety, which is exacerbated by poor animal-to-human translation and insufficient genetic evidence and biomarker data. The area is growing beyond small compounds to biologics (antibodies, peptides, siRNA, and gene therapy) and innovative modalities such as PROTACs to address intractable targets, albeit these techniques are more expensive. Key molecular challenges include achieving selectivity among closely related protein families, such as GPCRs, kinases, and phosphatases. Researchers also need to address protein flexibility in disordered targets that do not follow the "lock-and-key" model. Additionally, targeting non-protein substrates like DNA and RNA raises specificity concerns. Finally, designing drugs for challenging pockets, including protein-protein interactions and polar or flat binding sites, often requires large or complex molecules, covalent ligands, or allosteric targeting with help from computational methods [24].
IMPORTANCE:
Continuous drug discovery and development are critical* for meeting unmet medical needs, responding to new risks such as pandemics, overcoming resistance, and delivering safe, effective, and accessible medicines globally. [25, 26]
Unmet Medical Demands.
A key justification for ongoing R&D is the persisting gap between illness burden and accessible therapies, particularly in complicated situations (neurodegeneration, uncommon genetic disorders, refractory malignancies) when present alternatives are insufficient or lacking. Modern pipelines include target selection, validation, and iterative optimization, but average timeframes (about 11-13.5 years) and success percentages are low, emphasizing the need for innovation to transform biological discoveries into therapeutic benefits. Enhancing academic-industry networks and integrated infrastructure can decrease fragmentation, speed up candidate advancement, and better connect research with clinical needs. [25, 26]
Lessons From COVID-19
The COVID-19 pandemic compressed decades of methodology into months, proving that well-planned platforms, adaptive trial designs, and coordinated data sharing may provide antivirals and immunomodulators quickly while maintaining evidential standards. The reviews summarize how repurposing, structure-guided design, and hit-to-lead "sprints" enhanced small-molecule antivirals and defined stage-appropriate immunomodulation, while also revealing gaps in preclinical models and equitable access. To assure generalizable safety and efficacy, future preparation calls for pre-positioned tests, broad-spectrum antiviral libraries, regulatory playbooks for faster nonclinical and clinical paths, and inclusive trials. [27-29]
Antimicrobial Resistance
Antimicrobial resistance (AMR) jeopardizes decades of progress by reducing the efficacy of antibiotics, antifungals, and antiparasitics, making ordinary infections more difficult to treat while increasing mortality and costs. Contemporary studies advocate for a twin strategy of reinvigorating antibacterial pipelines and pursuing alternative techniques such as bacteriophages, lysins, potentiators, and immune modulators, as well as diagnostics-guided stewardship to reduce resistance selection. Frontline research focuses on next-generation options for diversifying modalities and reducing resistance pressure, including as host-directed therapy, antiviral medicines, and modified biologics. [30-32].
Personalization And Demographic Disparities
Drug response differs depending on age, gender, genetic background, and comorbidities, necessitating biomarker-guided development to match the appropriate medication to the right patient at the right dose. Precision-medicine frameworks built into discovery—companion diagnostics, pharmacogenomic enrichment, and adaptive designs—improve benefit-risk by anticipating variability in exposure and impact. These techniques also help with pediatric and geriatric formulations and dosage regimens, expanding therapeutic reach across the life cycle. [25, 26, 33]
Technology-Driven Acceleration
Advances in computational and experimental technology are altering the economics and timing of discovery. In-silico approaches, including as virtual screening, physics-informed docking, and AI-guided de novo design, prioritize chemical materials with a better possibility of success and speed up hit-to-lead selection. Reviews detail how AI/ML works with high-throughput screening, structure-based design, and systems pharmacology to minimize late-stage attrition, optimize ADME/Tox profiles earlier, and promote rational polypharmacology for complicated disorders. To guarantee generalizability and repeatability, these benefits must be supported by high-quality, interoperable datasets and transparent benchmarking. [26, 33, 34]
Clinical Assessment And Evidence Creation.
Despite advances in research, clinical trials remain the judge of safety and efficacy; technique innovation prioritizes speed while maintaining rigor. COVID-19 lessons highlight adaptable platform trials, smooth phase transitions, and pharmacokinetic/pharmacodynamic modeling to improve dosage windows and stage-appropriate therapies. Post-marketing surveillance and real-world evidence are then used to assess long-term safety, uncommon adverse events, and efficacy in varied groups, informing labeling revisions and lifecycle optimization. [27-29]
Economic And Social Value
Drug discovery requires a significant investment of resources, yet effective medications minimize hospitalizations, disability, and productivity loss, especially in high-burden illnesses and infections. Reviews advocate for novel incentive structures and collaborations to de-risk high-need regions (e.g., Gram-negative infections) and sustain pipelines when traditional market returns are low, therefore connecting innovation with public health objectives. Coordinated academic consortiums, public-private partnerships, and charitable and non-profit organizations may invest wisely in underserved areas while maintaining global access. [25, 26, 32]
Access, Equity, and Preparedness
Equitable access is critical to public health impact: technology transfer, tiered pricing, and quality-assured generics guarantee that scientific innovations benefit the whole population, especially in low- and middle-income nations. Pandemic-focused evaluations emphasize preparation architectures—priority pathogen portfolios, common procedures, and regulatory flexibilities—for moving from sequencing to first-in-human in weeks while retaining safety precautions. Integrating equity (inclusive eligibility, varied venues, and community participation) boosts representativeness and trust, resulting in more robust and generalizable data. [27-29]
Future Directions.
Priorities identified by authoritative surveys include expanding modality variety (small compounds, biologics, RNA, cell/gene treatments), fortifying antimicrobial pipelines with novel agents, and mainstreaming AI throughout design-make-test-analyze cycles with thorough validation. Improving translational models, data standards, and open-science cooperation will eliminate redundancy and speed up iterative learning across programs and illnesses. Achieving these aims connects scientific innovation to legislative frameworks that reward social value rather than unit sales, assuring sustainability for both urgent and neglected needs. [25, 33, 34]
CONVENTIONAL METHODS:
Random Screening
In the absence of recognized medications or other molecules with desired action, a random screen is a useful method. Random screening does not include intellectualization; all compounds are screened in the bioassay regardless of their structure. The two main kinds of materials tested are synthetic chemicals and natural products (microbial, plant, and marine) [35]. Plants for pharmacological screening can be chosen at random, with all plants from a certain region being chosen and screened for various activities. As a result, random screening does not require any good rationale in the literature. Plants can also be chosen for research if they are high in phytochemicals or if other members of the same family exhibit biological activity. The latter is because numerous related species exhibit comparable phytochemicals. Random screening and phytochemical approaches have been shown to be quite effective in discovering novel leads, particularly when folk knowledge is missing. These save many plants from being overlooked because of a lack of supporting information [36-38].
Ethnopharmacology
Ethnopharmacology, which was officially acknowledged in 1967, combines centuries of traditional plant-based knowledge with contemporary scientific methodologies to confirm and exploit the medicinal potential of natural products[39].By integrating ethnobotanical and ethnomedical literature with multidisciplinary collaboration between botanists, pharmacologists, and chemists [36,39,40]. It conducts systematic research on bioactive substances for safety, effectiveness, and commercial synthesis, therefore protecting cultural heritage and assisting herbal practitioners. Compared to random screening and phytochemical techniques, ethnopharmacology is more cost-effective and focused, eliminating resource waste and ensuring that potential species are not ignored. Despite obstacles such as restricted access to modern analytical tools in impoverished countries, this technique acts as an important link between traditional medicine and evidence-based medication development.
The Role Of Serendipity In Drug Discovery
Serendipity, defined as the ability to make unexpected, pleasant discoveries, has played a critical role in drug discovery, transforming random observations into significant therapeutic achievements. Early chemical discoveries fueled the growth of organic chemistry and the pharmaceutical sector, with dye companies such as Bayer and Ciba transitioning into medication producers. Penicillin's accidental discovery revolutionized antibiotics and ushered in a golden age of antimicrobial research, whereas benzodiazepines were discovered by chance and altered psychiatric medicine. Chance observations combined with rigorous investigation gave rise to monoamine oxidase inhibitors, laying the groundwork for neuropsychopharmacology, and in modern times, sildenafil's unintended erectile effects shifted its use from angina treatment to the blockbuster therapy for erectile dysfunction—demonstrating that, even today, "chance favors the prepared mind." [41]
Traditional drugs discovery
Traditional drug discovery has long been dominated by laborious, costly empirical screening of large compound libraries, limiting chemical exploration and frequently yielding serendipitous hits rather than designed leads; these methods offer little mechanistic insight into molecular interactions, making prediction of compound behavior in complex biological systems difficult and frequently overlooking critical pharmacokinetic and bioavailability factors that govern absorption, distribution Ethical and safety issues, notably over animal experimentation and hidden dangers, have posed further challenges to established procedures. Genomics, high-throughput screening, molecular biology, structural biology, bioinformatics, and computational approaches transformed the area in the late twentieth century, significantly speeding target discovery and lead optimization. Simultaneously, the long-standing influence of traditional medical knowledge—from ancient surgical methods to holistic health philosophies—continues to inform modern strategies, combining historical wisdom with cutting-edge technologies to create a comprehensive, mechanistic approach to lead discovery and candidate optimization [44, 45]. Traditional drug development is hampered by expensive costs and lengthy schedules, with many compounds failing in subsequent stages. It struggles to discover new molecules and efficiently treat complicated diseases such as cancer. Drug resistance, illness complexity, loss of natural resources, and limited chemical variety in libraries all impede development. These problems emphasize the need for new technologies such as artificial intelligence and bioinformatics to change drug research [46].
Stages Of Drug Discovery
Drug development is a sophisticated and well-structured process that converts scientific research into useful therapies. It has several steps, ranging from finding possible targets to testing and guaranteeing the safety and efficacy of novel drugs (Fig. 3) each step is essential in the development of medications to enhance health and treat disorders.

Figure 3: Sequential Stages Involved In Drug Discovery
1. Target identification
Target identification and validation are crucial components of current drug development, beginning with the identification of biological entities (genes, proteins, or signaling pathways) that play critical roles in disease pathogenesis and offer possible therapeutic intervention sites. Genome-wide association studies (GWAS) scan the genome for single-nucleotide polymorphisms (SNPs) associated with disease phenotypes, high-throughput genomic sequencing (whole-genome and whole-exome) reveals rare causal mutations in monogenic disorders, and advanced functional genomics techniques such as CRISPR-Cas9 screens and RNA interference to assess gene essentiality and map interaction networks. Integrative multi-omics techniques enhance target identification by combining transcriptomic, proteomic, and metabolomic data to create comprehensive disease landscapes and find critical regulatory nodes. Following target identification, rigorous validation confirms their causal role and druggability using complementary genetic methods such as CRISPR-mediated knockouts or knockins and RNAi knockdowns, pharmacological probing with inhibitors, activators, or allosteric modulators, and in vivo evaluation using knockout, transgenic, and xenograft animal models to assess efficacy and safety in physiologically relevant contexts. By prioritizing targets with strong biological underpinnings and demonstrable therapeutic modulation, these processes significantly reduce late-stage clinical trial failures, streamline development timelines, lower costs, and support precision medicine initiatives by allowing the design of patient-specific therapies based on validated molecular biomarkers [47].
2. Assay
Assay development is a critical stage in drug discovery because it converts a confirmed biological target into a dependable test system that analyzes how potential compounds interact with that target. Effective tests are classified into three complimentary types: cell-based, biochemical, and in silico. Cell-based assays employ living cells or sophisticated 3D cultures to record complex physiological responses such as signaling alterations, toxicity, and gene expression, allowing for spatial and temporal resolution of drug effects in a physiologically appropriate setting. Biochemical assays use purified targets to quantify binding affinities and kinetic parameters with high sensitivity and reproducibility. Techniques used include fluorescence resonance energy transfer (FRET), surface plasmon resonance (SPR), and enzyme-linked immunosorbent assays (ELISA). In silico assays use computational approaches like as molecular docking, ligand-based screening, and quantitative structure-activity relationship (QSAR) modeling to digitally screen millions of compounds, estimate ADMET characteristics, and rank candidates for laboratory testing. To ensure relevance, sensitivity, and reproducibility, a robust assay must be developed through thorough optimization and quality control. To identify actual hits from noise, key metrics such as the Z′ factor must routinely surpass thresholds (e.g., >0.4), and assay conditions—reagent concentrations, incubation periods, and solvent tolerances—must be properly adjusted to reduce artifacts. By combining data from cell-based, biochemical, and computational experiments, researchers may quickly evaluate large compound libraries, focus resources on the most promising findings, and reject inappropriate candidates. This comprehensive strategy speeds lead optimization, lowers late-stage failures, and eventually reduces development timeframes and costs, allowing for more efficient delivery of safer and more effective medications to patients [48].
3. HIT
In drug discovery, a hit compound is a molecule that demonstrates the required activity against a biological target in an initial compound screen and whose activity is verified during retesting. Hit identification encompasses various screening strategies, including high-throughput screening (HTS), which tests large compound libraries without prior structural knowledge; focused screening, which selects compounds based on knowledge of target interactions; fragment screening, which involves small molecular fragments screened at high concentrations with the aid of protein structure data; and physiological screening, which uses tissue-based assays to simulate in vivo effects more closely. Hits serve as the foundation for medicinal chemistry improvement to increase potency, selectivity, and drug-like qualities, and are then tested in cell-based disease models and animal investigations during lead discovery [54]. Academic institutions with industrial-standard chemical libraries and screening tools are increasingly contributing to hit identification, taking early drug development outside typical pharmaceutical settings. The thorough identification and validation of hits is essential for providing reliable chemical starting points for effective drug development [55].
4. Lead identification & optimization
Lead identification and optimization are critical steps of drug discovery that include choosing and refining molecules with desirable biological activity before developing them into clinical prospects. Lead identification uses techniques such as high-throughput screening (HTS), virtual screening, molecular docking, and machine learning to select compounds based on solubility, purity, metabolic stability, and other drug-like qualities. Lead optimization then improves these compounds' potency, selectivity, pharmacokinetics, and safety using methodologies such as functional group alteration, structure-activity relationship (SAR) investigations, and pharmacophore-based design. NMR, mass spectrometry, QSAR, and computer models (such as LEADOPT and CoMFA) help with structural refinement, whereas ADMET profiling and animal studies assess safety and effectiveness. To improve drug development speed and success rates, better toxicological testing, biomarker-based translational models, cost-effective miniaturized assays, and industry-academic collaborations are crucial [56].
5. Preclinical and Clinical Development in Drug Discovery
Preclinical testing is a vital step of drug development in which animal tests are performed to assess the safety, effectiveness, and possible dangers of a medication candidate before it is administered to people. These studies, conducted in accordance with ICH regulations, concentrate on pharmacology and toxicology: pharmacology evaluates pharmacokinetics (absorption, distribution, metabolism, and excretion) and pharmacodynamics to determine half-life, bioavailability, receptor affinity, and metabolic profile, whereas toxicology identifies harmful or unwanted effects using both in vitro and in vivo models. Because many medications might have species-specific effects, choosing the appropriate animal model is critical. Once enough data has been collected, developers must submit an Investigational New Drug (IND) application to the FDA before clinical trials can begin; this dossier includes preclinical results, toxicity data, manufacturing details, proposed clinical protocols, prior clinical data (if available), and information on the researcher or research institute. Only after IND approval can human clinical research begin, which is carried out in phased clinical trials designed to evaluate safety, dosage, efficacy, and long-term outcomes using protocol-defined criteria such as participant selection, route of administration, study duration, and data analysis methods. If clinical and preclinical results show that the drug is both safe and effective, the developer files a New Drug Application (NDA) that includes the drug's entire history, including study data, labeling proposals, directions for use, potential for abuse, safety updates, patent information, and compliance with institutional review standards. The FDA reviews the NDA for 6-10 months. If the data proves the drug's safety and efficacy, it is approved with finalized labeling to guide proper use and ensure safety. Otherwise, the NDA may be rejected or additional studies may be requested. Applicants have the right to appeal [57]. Clinical trials are critical phases in drug research in which novel medications are tested on humans to establish their safety, efficacy, and optimum dosage. These trials are undertaken in phases—Phase I, II, III, and IV—with distinct purposes, ranging from checking safety in healthy volunteers to monitoring long-term effects after the medicine is authorized. (Fig. 4)
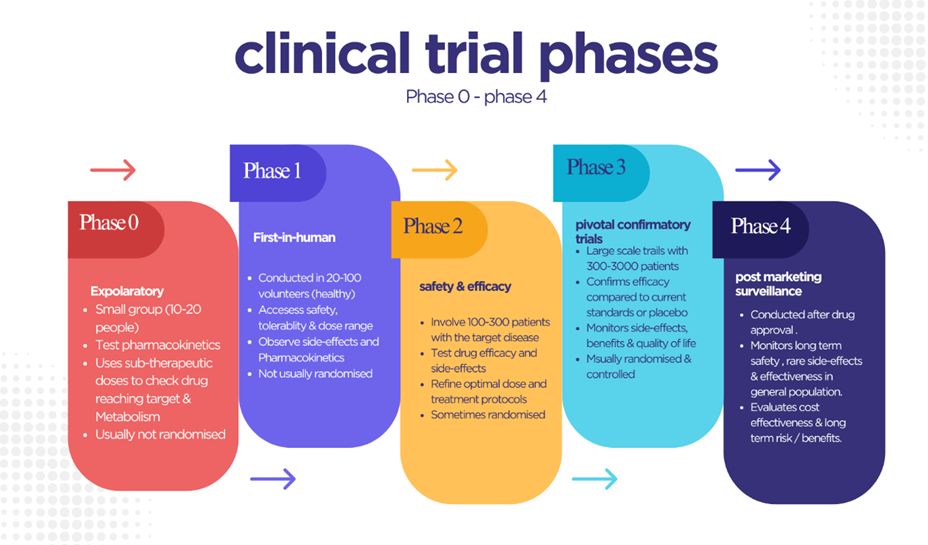
Figure 4: Phases of Clinical Trials in Drug Development
APPLICATIONS:
Brief Introduction To Rentosertib
Rentosertib (previously ISM001-055) is a watershed moment in pharmaceutical history since it is the first experimental medicine whose biological target and therapeutic component were found using generative artificial intelligence [58]. Rentosertib, developed by Insilico Medicine using their proprietary Pharma.AI platform, is a first-in-class TNIK (TRAF2- and NCK-interacting kinase) inhibitor designed to treat idiopathic pulmonary fibrosis (IPF), a severe progressive lung disease affecting approximately five million people globally with a median survival of only three to four years[58-60].The medication's development path from target identification to Phase 2a clinical trials was accomplished in less than 30 months, which is a remarkable timeframe when compared to standard drug discovery methods that generally take 10-15 years[58,59](Fig. 5). Rentosertib reached a crucial milestone in June 2025, when it demonstrated both excellent safety profiles and prospective efficacy in a Phase 2a clinical study published in Nature Medicine, becoming the first AI-discovered medicine to exhibit clinical proof-of-concept in humans[59-61].The applications listed below illustrate the unique AI-driven approaches that allowed Rentosertib's quick discovery and development, demonstrating how artificial intelligence is revolutionizing all stages of pharmaceutical research (Fig. 6).
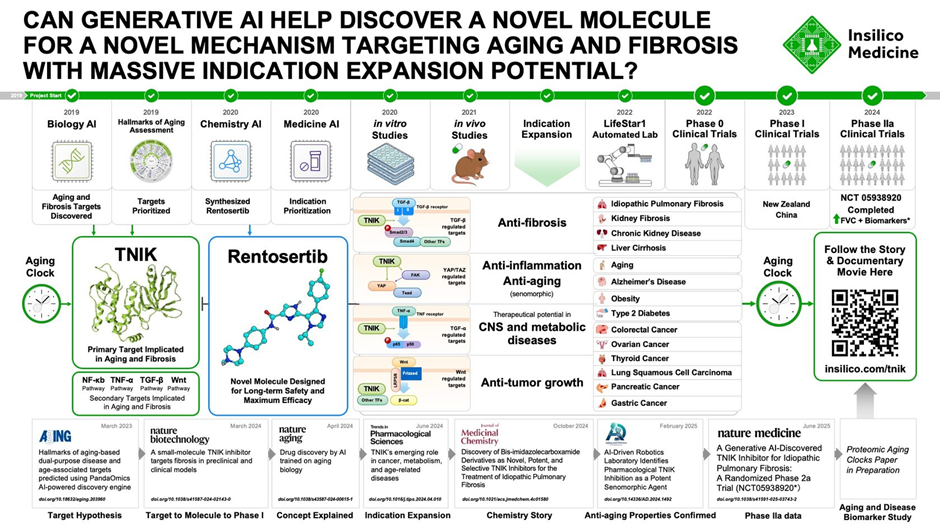
Figure 5: Generative AI–Driven Discovery of Rentosertib, Targeting Aging and Fibrosis, from Hypothesis to Clinical Trials
AI-Driven Drug Discovery Applications: The Rentosertib Methodology
1. AI-based Target Identification.
The Application: The PandaOmics platform from Insilico Medicine used ensemble machine learning techniques, transformers, and natural language processing to systematically examine large datasets such as genomes, transcriptomics, proteomics, and biomedical literature. The AI system evaluated hundreds of candidate pharmacological targets based on clinical relevance, route centrality, genetic evidence, and therapeutic feasibility [58, 59, 62].
Rentosertib Implementation: PandaOmics discovered TNIK as a unique therapeutic target for IPF research due to its essential role in directing profibrotic and proinflammatory cellular activities. This AI-driven target identification method selected TNIK from thousands of possible candidates, giving computational proof for its participation in fibrotic disease pathways before any experimental validation was performed [58, 59, 62].
2. Multi-Omics Data Integration and Validation
Advanced unsupervised learning algorithms and network embedding techniques use diverse biological datasets such as genomic aberrations, differential gene expression profiles, protein-protein interaction networks, and pathway analysis data to validate target relevance and mechanism [59, 62].
Rentosertib Implementation: After TNIK was found, AI models used multi-omics profiles to demonstrate its mechanistic significance in IPF pathogenesis. Before beginning wet-lab tests, the system verified TNIK's role in important fibrotic and inflammatory signaling cascades, giving strong computational evidence to support its potential as a disease-modifying target [59, 62].
3. Generative Molecular Design and De Novo Chemistry
The Application: Insilico's Chemistry42 platform uses generative deep learning techniques such as variational autoencoders (VAEs), generative adversarial networks (GANs), and reinforcement learning to create unique chemical compounds with desired biological features [58,63,64].
Rentosertib Implementation: Chemistry42 developed hundreds of unique small-molecule compounds that are particularly intended to inhibit TNIK. The AI approach optimized molecular structures for many characteristics at once: binding affinity to the TNIK kinase domain, selectivity against off-target kinases, drug-like physicochemical qualities (Lipinski compliance), and synthetic accessibility. Within weeks, this method generated around 80 distinct molecular candidates, each representing a completely novel chemical framework not found in current compound libraries [58, 62].
4. Structure-Based Virtual Screening and Affinity Prediction
The application uses graph neural networks and AI-enhanced molecular docking methods to predict binding interactions between small compounds and target proteins based on crystal structures and AlphaFold predictions. Multi-task learning models include binding affinity, selectivity, and ADMET characteristics into a single scoring function [65, 66].
Rentosertib Implementation: AI models compared all produced Rentosertib candidates to the TNIK kinase structure, predicting binding poses, affinity, and selectivity profiles. The method selected molecules with superior binding properties to TNIK's ATP-binding site while limiting interactions with related kinases, allowing for targeted experimental validation of just the most promising candidates [62].
5. Predictive ADMET and Pharmacokinetic Modeling
The Application: Deep neural networks trained on large pharmaceutical datasets predict drug candidates' absorption, distribution, metabolism, excretion, and toxicity (ADMET) characteristics prior to synthesis, allowing for early optimization of pharmacokinetic profiles[67,68].
Rentosertib implementation: During the design phase, artificial intelligence models projected the metabolic stability, hepatic clearance, probable medication interactions, and safety profiles of Rentosertib. These predictions influenced structural modifications to improve oral bioavailability and reduce toxic liabilities, which contributed to the drug's favorable pharmacokinetic profile in Phase 1 clinical trials, where it demonstrated appropriate exposure levels and tolerability in healthy volunteers [58, 62, 69].
6. Automated Lead Optimization
The Application: Bayesian optimization algorithms and reinforcement learning agents use iterative design cycles to balance several drug development objectives such as potency, selectivity, metabolic stability, solubility, and synthetic accessibility [66, 70].
Rentosertib Implementation: Multi-objective optimization aimed to balance TNIK inhibitory efficacy, kinase selectivity, oral bioavailability, and manufacturing feasibility. AI agents proposed minimum structural changes to improve the overall drug profile, allowing for quick optimization cycles that resulted in compounds with nanomolar potency against TNIK while keeping favourable development characteristics [62].
7. Clinical Trial Design and Biomarker Discovery
The Application: Machine learning models assess past clinical trial data, patient records, and biomarker profiles to optimize trial design, patient selection criteria, and endpoint definitions for greatest chance of success [59, 69].
Rentosertib Implementation: AI technologies assisted in the design of the Phase 2a GENESIS-IPF study by reviewing previous IPF studies to optimize patient inclusion criteria, dosing techniques, and trial objectives. The system identified relevant biomarkers for monitoring treatment response, including serum proteins associated with fibrosis (COL1A1, MMP10, FAP) and inflammation (IL-10), which were then validated in the clinical trial and showed dose-dependent changes correlating with clinical improvements. [59, 61, 69]
This comprehensive use of AI throughout the Rentosertib development process from initial target discovery to clinical validation shows how artificial intelligence can revolutionize pharmaceutical research by significantly reducing timelines, costs, and development risks while identifying novel therapeutic opportunities that would not have been discovered using traditional approaches.

Figure 6: Comparison of Traditional VS. AI-Driven Drug Discovery Approaches
ADVANTAGES OF AI IN RENTOSERTIB DISCOVERY:
Faster Target Identification And Validation
AI integrates multi-omics datasets, signaling networks, and disease context evidence to prioritize causal targets faster than manual hypothesis generation, enabling rapid movement from broad biological hypotheses to actionable targets with mechanistic plausibility. In idiopathic pulmonary fibrosis (IPF), this approach elevated TNIK and supported swift validation aligned with profibrotic pathways, establishing a solid foundation for rentosertib's program. [59, 71]
Generative, Directed De Novo Design.
Data-driven generative chemistry creates novel structures based on multiple criteria, including target potency, kinome selectivity, physicochemical constraints for oral exposure, and synthetic feasibility. Feedback loops with active learning efficiently shrink chemical space, resulting in a balanced TNIK inhibitor that progressed to a clinical candidate on compressed timelines in rentosertib's case. [59, 72]
Early ADMET Triage And Off-Target Risk Minimization.
Machine-learning ADMET models, trained on large toxicology and PK corpora, flag structural alerts (hepatotoxicity, genotoxicity, cardiotoxicity proxies) and predict metabolic liability early, guiding medicinal chemistry to mitigate risks while maintaining activity. In kinase programs, AI-assisted selectivity modeling reduces unintended kinome engagement and late-stage attrition, improving the probability of rentosertib translating safely to humans. [72, 73]
High-Content Phenotyping And Organ-On-Chip Analysis.
Deep-learning computer vision can quantify fibrosis-related changes (e.g., myofibroblast activation, matrix remodeling) across dose-time matrices with greater sensitivity and reproducibility. This improves exposure-response understanding and translational confidence for compounds like rentosertib. [73, 74]
Digitally Guided Clinical Design And Dosage Strategy.
Digital twin simulations, based on real patient distributions, allow for in-silico exploration of dosing schedules, endpoint sensitivity (e.g., FVC trajectories), and enrichment criteria, informing trial sizes and inclusion rules that preserve power and feasibility. This data-guided planning supported rentosertib's path into early trials and interrogation of dose-dependent lung function effects in phase 2a [59, 74]
Timeline Compression And Cost-Efficiency
Generative design prioritizes high-yield chemical space, predictive ADMET reduces attrition, and active-learning experiment selection reduces low-information studies. This results in fewer compounds synthesized and fewer dead-end experiments, allowing for earlier human data generation, as demonstrated by rentosertib's accelerated trajectory from target discovery to early clinical evaluation. [59, 72]
Precision Medicine Foundations And Biomarker Strategy
Machine learning identifies composite biomarkers (transcriptomic/proteomic signatures, imaging features) linked to response, allowing for stratified enrollment in mid-stage studies and personalized therapy. In IPF, aligning mechanistic TNIK inhibition with molecular responders provides a principled foundation to optimize rentosertib's clinical impact and design rational combinations that converge on complementary fibrotic pathways. [59, 71]
Better Judgments Under Uncertainty In R&D.
Active learning and Bayesian frameworks minimize uncertainty by selecting the optimal synthesis, assay, or dose step, preventing overconfidence in early positive signals and underreaction to weak safety concerns. This systematic approach to decision-making maintains speed and scientific discipline in programs like rentosertib. [72, 73]
End-To-End Translational Coherence
AI aligns the entire chain from systems biology target rationale (TNIK's role in fibrotic signalling) to molecular design (selective inhibition with developable properties), phenotypic linkage (anti-fibrotic signatures), and clinical endpoints (dose-dependent FVC trends), improving interpretability for investigators and regulators and increasing the likelihood that mechanistic insight persists through clinical complexity, as observed in the rentosertib study. [59, 71]
Institutional Learning And Reuse Intelligence
Rentosertib's discovery data improves default strategies, including starting chemotypes, safety trade-offs, and experiment sequencing, for future fibrosis and kinase programs, resulting in increased productivity over time. [72.73]
Scalability And Cross-Program Leveraging
Established AI workflows—target triage, generative policies, ADMET predictors, and digital trial simulator generalize across indications; insights from rentosertib's TNIK program transfer to related network nodes and kinase families, accelerating setup and reducing ramp time in new fibrosis or inflammatory projects. Human-machine cooperation increases rigor. [72, 74]
AI enhances rather than substitutes expert judgment: chemists evaluate synthetic realism, biologists ensure disease relevance, and doctors align endpoints with patient benefit. Rentosertib's growth exemplifies this partnership AI speeds and structures work, while domain knowledge maintains biological plausibility, clinical feasibility, and safety [59, 73].
DISADVANTAGES
Data Quality, Representativeness, And Bias.
AI systems in drug discovery are extremely sensitive to the breadth, quality, and representativeness of training data; gaps such as batch effects, label noise, inconsistent assay conditions, and skewed demographics can lead to biased or unstable models that overestimate efficacy or toxicity, particularly when extrapolating to new chemical matter or underrepresented patient populations. In the rentosertib context, any bias in fibrosis cohort data or omics sources used to prioritize TNIK or build predictive ADMET models could distort safety and response expectations, risk cohort imbalances and unanticipated adverse events when moving from controlled assumptions to heterogeneous real-world patients.Bias mitigation involves rigorous curation, augmentation, and continuous recalibration when new wet-lab and clinical data enter. However, these procedures add time, expense, and methodological complexity, which can reduce headline speed improvements claimed to AI [73].
Complex Models Are Opaque And Have Limited Interpretability.
Deep learning and ensemble systems frequently operate as black boxes, making it impossible to determine why a scaffold is prioritized, a target is ranked, or a toxicity alert is raised—posing a challenge for scientific hypothesis production, internal review, and regulatory assessment. Insufficient interpretability in discovery settings can complicate medicinal chemistry decisions (e.g., which functional group drives a predicted liability) and hinder mechanism-of-action refinement. In clinical planning, opaque response models may not provide the causal clarity needed for dose justification or enrichment strategies. Although explainable AI (XAI) techniques are improving, consensus standards for strong, quantitative explanation quality are still emerging, and additional explanation layers may create new failure modes or be inconclusive for highly nonlinear models. [75]
Overfitting, Generalization Gaps, And Domain Shift.
Models focused on specific chemotypes, assay formats, or site practices may not perform well when faced with domain transition, such as novel scaffolds, laboratories, or patient heterogeneity. This might result in optimistic discovery-stage metrics that do not translate into clinical outcomes. Rentosertib's journey shows that even with strong preclinical signals and model-guided design, clinical translation is still dependent on human biology's variability. Generalization gaps can emerge as safety signals or dampened efficacy outside the training distribution, necessitating conservative interpretation of in silico wins and rigorous prospective validation plans. [73]
Limited Understanding Of Safety Biology And Off-Target Consequences.
Although computational toxicology has evolved, it still suffers with rare idiosyncratic toxicities, metabolite-driven effects, and complicated, tissue-specific liabilities. Kinase programs can conceal tiny off-target engagements that avoid early predictors, appearing later as organ-specific toxicities. In rentosertib's phase 2a research, liver-related adverse events led to discontinuations, emphasizing the importance of rigorous wet-lab toxicology and clinical monitoring in AI-era pipelines. While in-silico filters reduce risk, they cannot replace empirical safety learning in humans. To avoid over-reliance, AI should prioritize safety science rather than replacing it entirely. [59, 73]
Privacy, Consent, and Governance for Clinical Data Use
AI's reliance on clinical narratives, imaging, and omics raises privacy, consent, and governance concerns regarding secondary use, re-identification risks, and cross-border data flows. Ethical deployment requires strong de-identification, data minimization, and explicit consent frameworks aligned with evolving regulations. Implementing these standards for trial planning or digital twin building can slow down AI-assisted trial operations due to additional procedures, audits, and documentation obligations. Rentosertib believes that aligning recruitment analytics with privacy rules is critical to maintaining trust and regulatory compliance as programs scale. [76]
Accountability And Liability Ambiguity
When AI-informed decisions lead to negative outcomes (e.g., dose selection based on algorithmic simulations), responsibility can be shared among model developers, sponsors, CROs, and investigators. However, current legal and regulatory frameworks offer limited guidance for assigning liability in hybrid human-AI decisions, potentially hindering innovation and complicating risk management. Clear governance rules, model risk management (MRM) techniques, and audit trails are required to attribute decisions and establish the evidentiary basis for design choices throughout the discovery and clinical stages [76]
Resource Concentration And The Digital Divide
Advanced AI requires specialized talent, large curated datasets, and high-performance compute. As a result, tools and outcomes can concentrate in well-resourced organizations, widening a digital divide that limits academic labs and smaller biotechs from fully participating. This may limit chemical diversity explored and independent validation. Proprietary models and non-public datasets limit reproducibility and external examination, hindering claims of faster timeframes and higher success rates. This dynamic may also affect downstream availability and affordability if innovation pipelines converge [72].
Regulatory Readiness And Evidence Standards.
Regulators expect transparent rationales for target selection, structure justification, preclinical model choice, and dose/exposure designs. Black-box predictions without mechanistic anchoring can slow reviews or trigger additional studies, offsetting some of AI's potential time savings. Credibility of first-in-class programs like rentosertib depends on traceable links from systems biology to molecular design and clinical endpoints. Building and documenting this chain requires significant effort in model validation, version control, and provenance tracking, which are still maturing in many discovery groups. [75]
Reproducibility, Provenance, And Auditability.
AI workflows might include several data transformations and model iterations; without strict data provenance, versioning, and audit trails, recreating discoveries becomes challenging, jeopardizing internal QA, cooperation, and regulatory inspections. Best practices—model cards, data sheets, experiment registries, and continuous performance monitoring—add process overhead but are required to ensure that enhanced velocity does not jeopardize scientific integrity or patient safety during translational processes. [72]
Human Expertise Loss And Automation Prejudice
If organizations rely too heavily on automated rankings or predictions, teams risk losing skills in medicinal chemistry intuition, pharmacology reasoning, and clinical trial craftsmanship; automation bias can allow weakly supported candidates or dosing choices to advance unchecked, especially when pressured to meet accelerated timelines. Maintaining a human-in-the-loop mentality, such as organized expert review, adversarial challenge sessions, and pre-mortems, balances algorithmic authority and ensures strong decision quality in systems like rentosertib. [73]
Excessive Optimism Might Pose Economic And Operational Problems.
Headline promises of timeline shortening can lead to under-resourced validation, optimistic trial powering, or premature scaling. However, real-world complexity (manufacturing, site variability, comorbidities) can cause delays and nullify initial AI-driven improvements. Longer-duration studies, broader geographies, and combination regimens for rentosertib will test AI-guided assumptions regarding dose, durability, and safety in the face of clinical heterogeneity, despite good phase 2a results. [59,72]
Security And Model Exploitation Threats
Model theft, data poisoning, or prompt/feature insertion can all compromise AI outputs, especially if external data streams are used for discovery or clinical analytics; such assaults could slightly bias target rankings or trial recommendations, endangering patient safety and exposing intellectual property. Implementing access limits, anomaly detection, and red-team reviews adds operational complexity to AI-enabled processes. [76]
To summarize, while AI has significantly expedited rentosertib's trajectory, the drawbacks revolve around data integrity and bias, interpretability and regulation, safety prediction limits, privacy and accountability, capability concentration, and operational maturity of AI governance. Programs that transparently handle risks, such as strong data stewardship, XAI, thorough wet-lab validation, human-in-the-loop decision protocols, and reproducible pipelines, are better positioned to transfer AI's speed into long-term, equitable patient benefits. [59, 72, 73, 75, 76]
Table.1: Summary of Advantages and Disadvantages of AI in Drug Discovery
|
ADVANTAGES |
DISADVANTAGES |
|
Faster target finding – AI helped spot TNIK quickly as a key fibrosis target. |
Data bias – If the data is incomplete or skewed, AI can give the wrong signals. |
|
Smarter drug design – AI can design better drug molecules faster than traditional methods. |
Black-box problem – Often, it’s unclear why AI made a particular choice |
|
Early safety checks – Predicts possible side effects before human trials |
Hidden risks – Some side effects still only show up in people, not models. |
|
Better clinical trials – Digital “patient twins” help decide doses and pick the right participants. |
Privacy & accountability – Patient data use and responsibility for mistakes are tricky issues |
|
Saves time and money – Cuts down on wasted experiments and speeds up testing |
Over-reliance – Too much trust in AI can weaken human judgment, plus rules and real-world hurdles slow things down. |
|
Personalized treatment – Helps match the right patients with the right therapy. |
Unequal access – Big pharma with money and tech benefits most, small labs may be left behind. |
|
Human + AI teamwork – Experts guide AI, keeping things realistic and safe Security |
Security risks – AI systems can be hacked or tampered with. |
FUTURE SCOPE:
Several significant developments in artificial intelligence (AI) have the potential to alter drug research. Quantum computing allows for far faster execution of complex molecular simulations, such as protein-ligand docking, than traditional approaches. Edge computing enhances this by processing data directly at laboratories or healthcare sites, lowering latency and allowing for real-time decision-making. Enhancements to explainable AI (XAI) are making previously opaque deep learning models more transparent. Chemical-explainable Graph Neural Networks (ChemXGNN) are used to determine the chemical bonds and functional groups that influence drug-target affinity predictions. This knowledge enables chemists to efficiently refine lead compounds, hence expediting optimization. Next-generation ADMET prediction platforms, such as admetSAR3.0, use large experimental datasets and advanced machine learning algorithms to increase the accuracy of absorption, distribution, metabolism, excretion, and toxicity predictions. A user-friendly interface offers simultaneous multi-endpoint analysis, allowing researchers to identify high-risk drugs earlier and decrease development expenditures. Federated learning tackles privacy and data silos by allowing institutions to train models jointly without sharing raw patient data. Personalized federated frameworks tailor models to specific patient subgroups or illness situations, facilitating the development of personalized therapeutics. Large language models (LLMs) are another frontier. LLMs speed up target identification and uncover novel disease-target relationships by swiftly mining enormous amounts of scientific literature. They also contribute to molecular design by recommending structural changes based on known activity patterns. In clinical development, LLMs can optimize trial protocols—recommending endpoints, sample sizes, and inclusion criteria—by learning from previous trial data, increasing trial efficiency and success rates. These developments, which include quantum and edge computing, XAI, sophisticated ADMET platforms, federated learning, and LLMs, will help to speed workflows, promote transparency, and protect data privacy. AI-driven drug discovery promises to bring safe, effective medicines to patients more quickly by shortening development timeframes and costs while enhancing prediction reliability [77].
SOLUTIONS TO OVERCOME AI CHALLENGES IN DRUG DISCOVERY:
Advanced data protection and privacy-preserving techniques, such as federated learning, enable institutions to train models on local datasets without disclosing sensitive patient information, thereby bridging data silos and maintaining confidentiality. Personalized federated learning improves models for specific patient subgroups, increasing predicted accuracy. To demystify “black box” AI, explainable AI (XAI) techniques like Chemical-explainable Graph Neural Networks include interpretability modules that uncover molecular factors driving predictions. This transparency fosters confidence and helps chemists optimize lead profiles. Reducing computational and environmental expenses requires efficient model design and hardware innovation. Optimized training algorithms and lightweight architectures use less energy, yet specialized AI accelerators, edge devices, and future quantum prototypes promise faster training with lower carbon footprints. Ensuring fairness necessitates bias identification and reduction during model development. . Data audits, reweighting or augmentation for underrepresented groups, and loss functions that include fairness all contribute to the prevention of discriminatory outcomes. Standardized curation and regulatory regimes ensure high-quality, representative datasets. Clear criteria for data collecting, pre-processing, and reporting, together with early coordination between businesses and regulatory agencies, guarantee compliance and robust verification strategies. Finally, multidisciplinary oversight by ethics committees, legal experts, and technologists is required to address informed consent, intellectual property, and cybersecurity. This integrated plan assures appropriate AI integration, which promotes patient safety and public trust [77].
CONCLUSION
From random screening to empirical plant-based remedies, the drug discovery method has developed to encompass biopharmaceutical discoveries, logical design, and, most recently, AI-powered discovery. Despite their significant contributions, traditional procedures are limited by their high costs, time commitments, and proclivity to fail in later clinical stages. Artificial intelligence gives solutions by enhancing lead optimization, lowering attrition, and increasing early prediction. As demonstrated by the Rentosertib instance, AI may accelerate and improve the accuracy of discovery, allowing for previously inconceivable milestones. Still, there are concerns about data quality, bias, model interpretability, regulatory challenges, and an overreliance on computer forecasts. As a result, AI should supplement rather than replace scientific judgment, experimental validation, and clinical competence. In the future, it is expected that merging AI with cutting-edge fields such as explainable AI, federated learning, and quantum computing would solve current challenges while increasing efficiency and transparency. To summarize, AI is a conceptual change rather than a technological development, and it is expected to play an important role in future medical research and discoveries. By merging human expertise and computer intelligence, the pharmaceutical business may enhance global public health by providing safer, quicker, and more personalized drugs [77].
REFERENCES





Tripthi Hinger, Shrutika Patil, Christina Viju, Jitendra Prajapati, Tanu Hinger, Rentosertib: An AI-Created Milestone Beyond Traditional Methods, Int. J. of Pharm. Sci., 2025, Vol 3, Issue 12, 1146-1170. https://doi.org/10.5281/zenodo.17840198
 10.5281/zenodo.17840198
10.5281/zenodo.17840198