
We use cookies to ensure our website works properly and to personalise your experience. Cookies policy
Abhilashi College of pharmacy, Nerchowk, Mandi Himanchal Pradesh175021, India
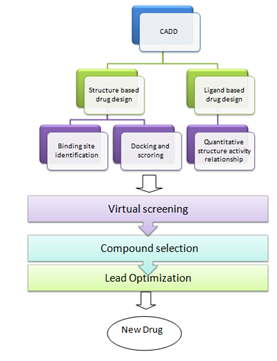
When combined with modern chemical biology screening techniques, computer assisted drug design is a potent tool for finding viable drug candidates. Despite the limitations and approximations it employs, CADD has emerged as a crucial step in the drug design process because of its capacity to expedite the discovery of new drugs by drawing on existing knowledge and theories about receptor-ligand interactions, energy and structural optimization, and synthesis. However, there is still potential for advancements, particularly with regard to scoring functions, taking into account solvent and molecular flexibility effects, focusing on receptors with little to no structural information, and enhancing computational effectiveness. Continuous advancements in the fields of chemical and structural biology, bioinformatics, and computer technology are required to improve current in silico methods. This will help to address the numerous drawbacks of virtual drug discovery techniques and enable computational drug design to reach its full potential.
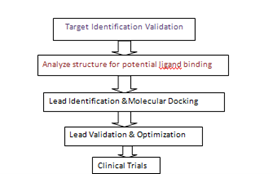
It takes a lot of money, time, and labour to introduce a new medicine to the market. Drug research and discovery takes an average of 10 to 15 years. High-throughput screening (HTS) and drug discovery have both benefited from advances in combinatorial chemistry that have increased the number of compound databases covering broad chemical spaces . Despite this, fewer novel molecular entities (NMEs) have been effectively introduced into the market during the past few years Rational drug design, as used in CADD, is a knowledge-driven technique that can produce important information regarding the binding affinity as well as the interaction pattern between protein and ligand (complex). Additionally, the availability of parallel computing, supercomputers, and specialised software has significantly accelerated the rate of lead identification in pharmaceutical research. The contemporary drug discovery scenario comprises a number of fields, including chemical and structural pharmacology, organic synthesis, computational chemistry, and biology. As a result, it is divided into numerous stages: (a) Target identification entails the finding and isolation of unique targets in order to research their roles and connections to a particular disease Target validation is the process of establishing a connection between the therapeutic target and the desired disease as well as the target's ability to control biological processes in the body after attaching to a partner molecule. To confirm that the target macromolecule is connected to the sick condition, numerous research are carried out2.
STRUCTURE-BASED DRUG DESIGN (SBDD)
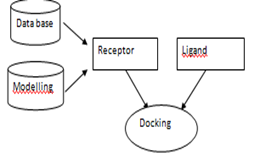
In SBDD, ligands are designed and evaluated based on their expected interactions with the protein binding site. This information is obtained from the binding site of a 3D macromolecule structure . Therefore, the first crucial steps in SBDD are the selection of a reliable pharmacological target and the gathering of its structural data. The development of protein structures was supported by structural and computational biology research using X-ray crystallography, nuclear magnetic resonance (NMR), cryo-electron microscopy (EM), homology modelling, and other techniques and simulations of molecular dynamics (MD) . According to , SBDD can be categorised into two categories:de novo strategy and virtual screening strategy. De novo drug design uses the 3D receptor's information to locate tiny pieces that are compatible with the binding site. To ensure synthetic accessibility, these fragments should be connected based on connection rules, resulting in a structurally unique ligand that may be synthesised for additional screening . . In contrast, virtual screening (VS) uses small molecule libraries that are already in existenc to find compounds with particular bioactivity that can be used to replace the ligands of target biomolecules or to find compounds for unexplored known targets with the help of structural data
DOCKING AND SCORING
One of the most well-known SBDD techniques is molecular docking, which predicts a compound's potential binding modes in a specific target binding site and calculates affinity based on conformation and complementarity with the features in the binding pocket . Virtual screening requires precise assessment of the binding and ranking of docked molecules. According to various article there are three categories of frequently used scoring functions:
LIGAND-BASED DRUG DESIGN (LBDD)
When the target protein's 3D structure is unknown, information gleaned from a collection of ligands active against a pertinent target (receptor or enzyme) can be used to pinpoint important structural and physicochemical characteristics (molecular descriptors) inferred to be in charge of the observed biological activity. , it is assumed that structurally related substances have similar biological responses and interactions with targets. To create a trustworthy ligand-based screening model, the compound set must cover a wide concentration range (at least 4 orders of magnitude). Quantitative structure-activity relationships (QSARs) and pharmacophore-based approaches are typical ligand-based design strategies.
QUANTITATIVE STRUCTURE–ACTIVITY RELATIONSHIP (QSAR)
The foundation of QSAR investigations is the idea that structural and molecular variations within a group of chemicals are related to changes in bioactivit. In order to design and mathematically forecast the biological property of novel chemicals, a statistical model is generated from this correlation. To create a trustworthy QSAR model, a number of restrictions must be followed: The training and test sets should be properly chosen, the ligands' molecular descriptors should not have any autocorrelation in order to prevent over-fitting, and there should be a sufficient amount of bioactivity data (minimum of 20 active compounds) collected using a common experimental protocol. (d) The model should be tested for applicability and predictability utilising internal and/or external validation . Some of the well-known applications or web servers frequently used for QSAR experiments are included in One of the most popular 3D-QSAR techniques is still Comparative Molecular Field Analysis (CoMFA) which was developed more than three decades ago.Spectral structure activity relationship (S-SAR), Topomer CoMFA , adaptation of the fields for molecular comparison (AFMoC) , and comparative residue interaction analysis are some more recent 3D-QSAR strategies. Despite its great achievements in the realm of drug discovery, More sophisticated multi-dimensional QSAR techniques, such as 4D, 5D, and 6D-QSAR, can address many of the problems that 3D-QSAR still has. While 5D-QSAR takes into account concerns like receptor flexibility and induced fit effects, 4D-QSAR was developed to address ligand conformation and orientation in the target binding region. In addition, 6D-QSAR recognises the importance of solvation effects in the interaction between receptors and ligands ( Through the use of Auto QSAR and Discovery Bus improvements in computational efficiency and software performance have also been made to QSAR model development and validation. By continuously incorporating new machine learning techniques, hundreds of highly predictive statistical models can be objectively found, updated, and validated in both approaches .
PHARMACOPHORE MODELING
In order to find molecules with various scaffolds but a comparable 3D layout of important interacting functional groups, pharmacophore screening is used. Utilising the bioactive conformation of potential drugs, binding site details can be added to the pharmacophore model.In QSAR modelling investigations, pharmacophore modelling is frequently carried out during the olecular alignment stage as well There are several widely used programmes for The programmes Discovery Studio, PHASE and LigandScout all use automatic pharmacophore creation.as well as MOE . Numerous reviews of these programmes and other algorithms have previously been published In order to decrease false negative and false positive outcomes, it is crucial to create a model with well-balanced sensitivity and specificity. To prevent making the model overly constraining, spatial limitations might be used in areas occupied by inactive chemicals and refined. Additionally, elements of the model that are not regularly shown in active compounds should be made optional or eliminated. To assess a model's sensitivity and specificity against an external test set following model refinement, validation experiments must be carried out .
INTEGRATION OF DRUG DISCOVERY TOOLS
Virtual screening does not come without its own drawbacks, despite its apparent benefits. It could be required to incorporate both LBDD and SBDD in order to meet all the practical requirements for finding a promising lead. In order to get over the drawbacks of either strategy, it is also possible to create a productive drug development process by combining virtual screening with HTS techniques. Numerous integration workflows exist, some of which are as follows: (a) Parallel integration involves either using several virtual screening protocols concurrently before HTS and experimental validation or using both virtual screening and HTS protocols concurrently. Since it has frequently been found that various hits (from different chemical spaces) are produced from different virtual screening techniques as well as from HTS , this strategy can offer enriched hit rates. Due to the extensive list of chemicals that can be created using this method, it may not be suitable for all studies. In order to continuously increase the selectivity of the screening operation, iterative or sequential integration combines computational and experimental approaches. Prior to experimental evaluation, the number of chemicals is reduced by sequential virtual screening approaches, which employ consecutive filters , Drwal and Griffith Visual inspection (which includes ligand binding, conformation at the active site, or the shape of the pharmacophore) and compound selection are primarily used in this final step prior to biological testing of a molecule. Another method involves experimentally validating virtual hits before using the data from in vitro screening to improve the in silico model This should result in stronger hits against the selected target. (c) Targeted integration creates a targeted library for experimental screening by pre-filtering compounds that are incompatible and unfavourable using computational approaches. Following the incorporation of virtual screening tools, a number of additional techniques, including approaches based on interactions, pharmacophores, and similarities, can be used. Incorporating computational drug design tools at any level of the drug discovery process unquestionably enables great information evolution that may result in better and more appealing drug prospects .
REFERENCES

Kiran Kumari, MuskanThakur, Sahil Dhiman, Shubham Dhiman, Role Of Computer Aided Drug Design In Modern Drug Discovery, Int. J. of Pharm. Sci., 2024, Vol 2, Issue 5, 157-162. https://doi.org/10.5281/zenodo.11114895
 10.5281/zenodo.11114895
10.5281/zenodo.11114895