Faculty Of Pharmacy, Bharath Institute Of Higher Education And Research
Alzheimer's disease (AD) is a complex neurodegenerative disorder characterized by progressive cognitive decline and memory impairment. Despite extensive research, current therapeutic options remain limited, and there is no cure. Recent advancements have expanded the understanding of key molecular mechanisms underlying AD pathogenesis, paving the way for novel therapeutic approaches. This review aims to provide a comprehensive analysis of the current drug targets for AD, focusing on various receptors, enzymes, and neurotransmitters involved in disease progression. The amyloid-beta (A?) cascade, tau protein hyperphosphorylation, synaptic dysfunction, oxidative stress, neuroinflammation, and impaired neurotransmission represent major pathological hallmarks of AD. Therapeutic strategies have been developed to target these processes, including ?-secretase (BACE) and ?-secretase inhibitors for reducing amyloid deposition, tau kinase inhibitors to prevent tau hyperphosphorylation, and cholinesterase inhibitors to enhance cholinergic neurotransmission. Additionally, drugs targeting N-methyl-D-aspartate (NMDA) receptors aim to prevent excitotoxicity. Emerging therapies focus on modulating neuroinflammatory pathways, targeting microglial activation, and blocking pro-inflammatory cytokines. Receptors such as nicotinic acetylcholine receptors (nAChRs), GABA receptors, serotonin receptors, and muscarinic receptors are explored for their roles in maintaining neurotransmitter balance and neuroprotection. This review will explore these drug targets in detail, discussing both established and experimental therapeutic strategies. We will provide illustrative diagrams and tables to highlight the receptor-ligand interactions, signaling pathways, and the role of various enzymes and neurotransmitters in the progression of AD. Additionally, we will discuss the clinical efficacy and challenges of current and potential
Alzheimer’s disease (AD) is a progressive neurodegenerative disorder characterized by cognitive decline, memory loss, and behavioural changes, primarily affecting the elderly. The exact etiology of AD remains complex, with multiple biochemical and genetic factors contributing to its pathology. Currently, available therapies offer symptomatic relief but fail to halt or reverse disease progression. Recent advances in understanding AD pathogenesis have identified several drug targets, including receptors, enzymes, and neurotransmitters involved in amyloid-beta (A?) plaque deposition, tau hyperphosphorylation, and neurotransmitter imbalances.[1] This review provides a comprehensive analysis of the key molecular targets in Alzheimer’s disease therapy, focusing on various receptors (such as NMDA, nicotinic, and muscarinic receptors), enzymes (like beta-secretase and gamma-secretase), and neurotransmitters (such as acetylcholine, glutamate, and serotonin). By exploring their roles in AD pathology and therapeutic potential, this review examines the challenges and advancements in drug design and clinical application. Additionally, emerging therapeutic approaches, such as immunotherapies, metal ion modulators, and neuroprotective agents, are discussed, emphasizing the future directions in AD drug discovery.[2]
Key Drug Targets in Alzheimer’s Disease
Alzheimer’s disease is associated with complex pathophysiological mechanisms, offering several potential drug targets. These targets are classified into receptors, enzymes, and neurotransmitter systems:
1. Receptors as Drug Targets
Several receptor systems are implicated in AD, and their modulation has shown potential in therapeutic interventions.[3]
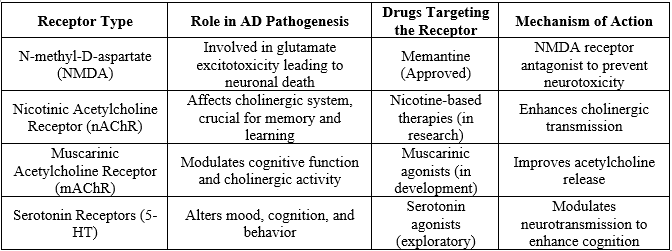
2. Enzymes Involved in Alzheimer's Pathology
The regulation of amyloid-beta (A?) production and tau phosphorylation is dependent on several key enzymes, making them attractive drug targets.
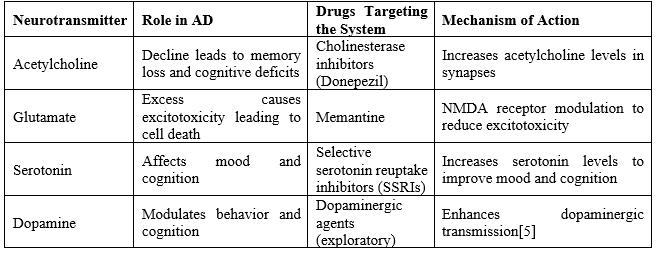
3. Neurotransmitter Systems in AD
Neurotransmitter imbalance contributes to cognitive decline in AD. Modulation of these systems remains a cornerstone of AD therapy.
1. Tau Phosphorylation Mechanism
In Alzheimer's disease, tau proteins, which normally stabilize microtubules in neurons, undergo abnormal phosphorylation. Hyperphosphorylated tau detaches from microtubules, aggregates into paired helical filaments (PHFs), and eventually forms neurofibrillary tangles (NFTs), contributing to neuronal dysfunction.
Key Enzymes Involved in Tau Phosphorylation:

Next, I’ll create a diagram illustrating the tau phosphorylation pathway.[5]
2. Neurotransmitter Receptor Subtypes
The neurotransmitter systems most implicated in Alzheimer’s are acetylcholine, glutamate, and serotonin, which are modulated through specific receptor subtypes.
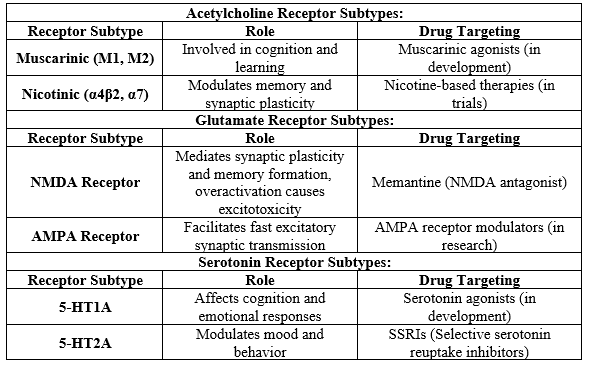
Next, I will create a diagram illustrating receptor subtype modulation in Alzheimer's.[6]
3. Drug Mechanisms in Alzheimer's Disease
In Alzheimer’s, drug mechanisms are categorized into several classes based on how they target the pathophysiological features of the disease:
Cholinesterase Inhibitors:
Donepezil, Rivastigmine, Galantamine: Inhibit acetylcholinesterase, increasing acetylcholine levels to improve memory and cognition.[7]
NMDA Receptor Antagonists:
Memantine: Prevents glutamate-induced excitotoxicity by modulating NMDA receptor activity.
BACE1 and Gamma-secretase Inhibitors:
Verubecestat, Semagacestat: Inhibit the enzymes responsible for amyloid-beta plaque formation (though clinical trials have seen limited success).
Tau Phosphorylation Inhibitors:
Tideglusib:
Inhibits GSK-3? to reduce hyperphosphorylation of tau proteins and prevent neurofibrillary tangle formation.
Tau is a protein responsible for stabilizing microtubules, which are crucial for the structural integrity and function of neurons. In Alzheimer's disease, abnormal hyperphosphorylation of tau leads to the formation of neurofibrillary tangles (NFTs), one of the hallmarks of the disease.[8]
Mechanism of Tau Phosphorylation:
The dysregulation of neurotransmitter systems is a key contributor to the cognitive deficits seen in AD. The most affected neurotransmitter systems include acetylcholine, glutamate, and serotonin. Let’s explore their receptor subtypes.
Acetylcholine Receptors:
Muscarinic Receptors (M1, M2):
These are G-protein-coupled receptors involved in various cognitive functions, including memory and learning.
Nicotinic Receptors (?4?2, ?7):
These receptors play a critical role in synaptic plasticity and memory formation.
Glutamate Receptors:
NMDA Receptors:
AMPA Receptors:
Serotonin Receptors:
3. Drug Mechanisms in Alzheimer's Disease
Different drug classes target the biochemical pathways involved in AD, focusing on reducing cognitive decline and preventing further neuronal damage. Here are the major drug classes
Cholinesterase Inhibitors:
NMDA Receptor Antagonists:
Memantine is the only approved NMDA receptor antagonist for moderate to severe AD. It works by blocking excessive NMDA receptor activation caused by high levels of glutamate, thereby preventing excitotoxicity and protecting neurons from damage. [17]
BACE1 and Gamma-secretase Inhibitors:
Tau Phosphorylation Inhibitors:
Tideglusib, a GSK-3? inhibitor, aims to reduce the hyperphosphorylation of tau proteins, preventing the formation of NFTs. This drug showed some promise in early trials, though further research is needed to establish its long-term efficacy.
Tau Protein:
GSK-3? and CDK5:
Neurofibrillary Tangles (NFTs):
Therapeutic Targeting:
2. Neurotransmitter Receptor Subtypes: Detailed Insights
Acetylcholine Receptors in Alzheimer's:
The cholinergic hypothesis proposes that a loss of cholinergic neurons in the basal forebrain contributes to the cognitive decline observed in Alzheimer's disease.
Muscarinic Receptors:
Nicotinic Receptors:
Glutamate Receptors in Alzheimer's:
Glutamate excitotoxicity:
In AD, excessive glutamate overstimulates NMDA receptors, leading to calcium influx, which triggers apoptosis (programmed cell death).
AMPA Receptors:
AMPA receptors are also involved in synaptic transmission. Dysregulation of these receptors in AD can impair synaptic plasticity and learning. AMPA modulators are in experimental stages to enhance synaptic function and memory.
Serotonin Receptors in Alzheimer's:
These receptors modulate mood and cognitive functions. Agonists of 5-HT1A may reduce neuroinflammation and improve cognition.
These receptors play a role in mood regulation and are targeted by antidepressants, such as SSRIs (Selective Serotonin Reuptake Inhibitors), which are commonly prescribed to AD patients for managing depression and anxiety.[23]
3. Drug Mechanisms: Expanded Details
Cholinesterase Inhibitors:
Mechanism:
Cholinesterase inhibitors work by blocking the breakdown of acetylcholine by acetylcholinesterase (AChE) in the synaptic cleft, thereby increasing the availability of acetylcholine for neurotransmission.
Donepezil:
Widely prescribed for all stages of AD, donepezil is a reversible inhibitor of AChE and helps improve cognitive symptoms.
Rivastigmine:
A dual inhibitor of AChE and butyrylcholinesterase (BuChE), rivastigmine is used for mild to moderate AD and is also available as a transdermal patch.
Galantamine:
In addition to AChE inhibition, galantamine acts as an allosteric modulator of nicotinic receptors, enhancing cholinergic transmission.[24]
NMDA Receptor Antagonists:
Memantine:
Memantine selectively blocks pathological overactivation of NMDA receptors without affecting normal receptor activity, thereby reducing glutamate-induced excitotoxicity.
BACE1 and Gamma-secretase Inhibitors:
BACE1 Inhibitors:
These drugs aim to prevent the formation of amyloid-beta (A?) by inhibiting the BACE1 enzyme that initiates the cleavage of amyloid precursor protein (APP).
Gamma-secretase Inhibitors:
These drugs target the enzyme that produces amyloid-beta after BACE1 cleavage. However, gamma-secretase also processes other important proteins, leading to adverse effects, such as gastrointestinal problems and skin disorders, resulting in discontinuation of many trials.
Tau Phosphorylation Inhibitors:
Tideglusib:
By inhibiting GSK-3?, this drug prevents the abnormal hyperphosphorylation of tau. Although it showed potential in early trials, further research is needed to optimize its efficacy and minimize side effects.[26]
1. Tau Phosphorylation Pathway: Clarification and Future Directions
Tau pathology is a key driver of neurodegeneration in Alzheimer's disease. The following additional points and emerging therapies focus on new strategies to modulate tau:
New Therapeutic Directions for Tau:
Anti-tau Antibodies:
One of the promising approaches involves using monoclonal antibodies to target pathological tau and prevent its spread. Clinical trials, such as semorinemab (anti-tau antibody), are ongoing to test their efficacy in reducing tau aggregation.
Tau Vaccines:
These vaccines aim to stimulate the immune system to clear pathological tau from the brain. Early clinical trials have shown that tau-targeting vaccines may reduce tau burden in animal models, and human trials are underway.[28]
2. Neurotransmitter Receptor Subtypes in Alzheimer's: Clarification and Potential Developments
Acetylcholine Modulation
The Decline of Cholinergic Neurons:
In AD, basal forebrain cholinergic neurons are severely affected, resulting in reduced levels of acetylcholine. The cholinergic hypothesis postulates that this loss leads to the hallmark symptoms of memory loss and cognitive decline.
Muscarinic Receptor Agonists:
AF102B and Xanomeline:
These are selective M1 muscarinic receptor agonists that aim to restore cognitive function by enhancing cholinergic transmission. Early trials with xanomeline showed promise in improving cognitive symptoms in AD patients, although side effects like nausea and vomiting have been a challenge.[29]
Glutamate Modulation:
Excitotoxicity:
The overactivation of NMDA receptors by glutamate leads to an influx of calcium into neurons, triggering cell death pathways. Preventing this overactivation without disrupting normal glutamate signaling is key to preserving cognitive function.
Potential NMDA Modulators:
SAGE-718:
A drug that modulates NMDA receptor activity, currently being studied for its potential to improve cognitive function in early Alzheimer's. It aims to enhance NMDA function selectively without causing excitotoxicity.[30]
AMPA Receptor Modulators:
Enhancing AMPA receptor activity may boost synaptic transmission and cognitive function. Drugs like CX-516 (AMPAkine) are under investigation for their ability to improve cognitive outcomes by facilitating synaptic plasticity.
Serotonin Modulation
5-HT1A Agonists: These drugs are being studied for their ability to modulate cognitive processes and mood. Serotonin reuptake inhibitors are already commonly used to manage depression and anxiety in AD patients, but selective serotonin modulators might enhance cognitive benefits.
3. Drug Mechanisms in Alzheimer’s Disease: Additional Clarifications and Future Prospects
Cholinesterase Inhibitors:
Future Developments:
Researchers are exploring next-generation cholinesterase inhibitors that are more selective or have fewer side effects than current drugs like donepezil. New delivery methods, such as transdermal patches, are also being studied to reduce gastrointestinal side effects commonly seen with oral medications.[31]
NMDA Receptor Antagonists:
Memantine's Success:
BACE1 Inhibitors:
Why Clinical Trials Struggled:
The failure of drugs like verubecestat in BACE1 inhibition trials stemmed from their lack of efficacy and high levels of adverse effects, including worsening cognition. This suggests that targeting amyloid-beta production alone may not be sufficient to halt disease progression.
Rethinking Amyloid Pathway Targeting:
Research is now focusing on more selective BACE1 inhibitors that can modulate amyloid-beta production without completely shutting down the pathway, thus minimizing off-target effects.[33]
Gamma-secretase Inhibitors:
Gamma-secretase Modulators (GSMs):
These modulate gamma-secretase activity without fully inhibiting it, aiming to reduce amyloid-beta production while avoiding side effects. GSMs are seen as a more promising approach compared to earlier gamma-secretase inhibitors, which had systemic toxicity.
Tau-targeting Therapies:
Ongoing Trials:
In addition to tideglusib, several other tau-targeting drugs are in trials:
Zagotenemab:
Another anti-tau monoclonal antibody that targets extracellular tau to prevent its spread between neurons.
TRx0237 (LMTX):
This drug is thought to disassemble tau tangles and prevent their aggregation, potentially reducing tau pathology.
Emerging Concepts in Alzheimer's Drug Development
Targeting Neuroinflammation:
Inflammation in AD:
Restoring Synaptic Plasticity:
Synaptic dysfunction occurs early in Alzheimer's disease, leading to cognitive deficits. Therapies aimed at enhancing synaptic plasticity, such as BDNF (brain-derived neurotrophic factor) mimetics, are being developed to restore neuronal communication and memory function.
LM22A-4, a small molecule that mimics BDNF signaling, is currently in preclinical testing for its potential to restore synaptic plasticity.[35]
Gene Therapy and CRISPR:
Gene-editing technologies like CRISPR-Cas9 are being explored to target genetic mutations (e.g., in APOE4) that increase Alzheimer’s disease risk. While still experimental, gene therapy holds long-term potential for halting or even reversing neurodegeneration.
1. Neuroinflammation in Alzheimer’s Disease
Chronic Neuroinflammation is increasingly recognized as a major contributor to Alzheimer's pathology. In the brain, inflammation is primarily mediated by microglia, the brain’s resident immune cells. While microglia help clear amyloid-beta deposits in early AD, chronic activation leads to the release of pro-inflammatory cytokines, contributing to neurodegeneration.[36]
Microglial Activation and Therapeutic Approaches
Microglia play a dual role: protective early on by clearing amyloid-beta but harmful as the disease progresses, contributing to synaptic loss and tau propagation.
Therapies Targeting Microglia:
Neflamapimod:
A p38 MAPK inhibitor that aims to reduce neuroinflammatory signaling in microglia. By inhibiting this pathway, neflamapimod could reduce synaptic dysfunction and improve cognitive outcomes. Clinical trials have shown it reduces inflammation and may slow memory loss.[37]
AL002:
This monoclonal antibody targets the TREM2 receptor on microglia, which plays a crucial role in the microglial response to amyloid plaques. Early studies suggest that enhancing TREM2 activity could promote the clearance of amyloid-beta and reduce inflammation.
Role of Astrocytes in Inflammation:
Astrocytes also play a role in Alzheimer's neuroinflammation. They become reactive and produce inflammatory molecules, which exacerbate synaptic damage. Therapies targeting astrocytes are still in the exploratory phase but could offer additional routes for managing inflammation.[38]
A Focus on BDNF and Neurotrophic Factors
Synaptic plasticity—the ability of synapses to strengthen or weaken over time—is crucial for learning and memory. In AD, synapses are damaged early, long before significant neuron loss. Enhancing synaptic plasticity could improve cognitive function, even in the presence of amyloid-beta and tau pathology.
Brain-Derived Neurotrophic Factor (BDNF) and Mimetics
BDNF is a neurotrophic factor that supports the survival of existing neurons and encourages the growth and differentiation of new neurons and synapses. In Alzheimer's, BDNF levels are reduced, which contributes to cognitive decline.
BDNF-Based Therapeutics:
LM22A-4:
A small molecule that mimics the action of BDNF by selectively activating its receptor, TrkB. By stimulating TrkB, LM22A-4 promotes synaptic plasticity and may reverse cognitive deficits in AD. Early studies in animal models show improved memory and synaptic function.
AAV-BDNF Gene Therapy:
This therapy delivers BDNF directly to affected brain regions using an adeno-associated virus (AAV) vector. Early trials in animal models have shown that increasing BDNF expression improves memory and reduces neuron loss, providing hope for its potential in human clinical trials.[39]
3. Gene Therapy and CRISPR in Alzheimer’s Disease
Gene therapy is emerging as a potential solution for targeting specific genetic risk factors associated with Alzheimer’s disease, particularly for individuals carrying high-risk mutations like APOE4. [40]
APOE4 and Gene Editing
APOE4 is the most significant genetic risk factor for Alzheimer's, increasing the risk of developing the disease by 3- to 15-fold compared to those carrying the APOE3 variant.
CRISPR-based Gene Therapy:
CRISPR-Cas9 gene-editing technology is being explored to modify the APOE4 gene and convert it into the less harmful APOE2 or APOE3 forms. Early research in animal models shows that CRISPR can successfully reduce APOE4 production, potentially lowering the risk of amyloid plaque formation.
AAV-APOE2 Therapy:
In this therapy, a virus is used to introduce the protective APOE2 gene into the brain. This approach is being studied as a potential treatment for individuals with the APOE4 gene to reduce their risk of developing Alzheimer's.[41]
4. Drug Trials and Future Prospects
Several clinical trials are underway, exploring new compounds and mechanisms that aim to slow or halt Alzheimer's progression. Here's an update on some of the most advanced and promising drug candidates:
Aducanumab and Anti-Amyloid Antibodies
Aducanumab (approved in 2021) was the first FDA-approved drug to target amyloid-beta directly. However, its approval was controversial due to mixed trial results. While it effectively clears amyloid plaques, its impact on cognitive decline remains unclear.
Lecanemab:
Another anti-amyloid antibody currently in phase III trials. Preliminary results suggest that it reduces amyloid plaques and shows more promise than aducanumab in slowing cognitive decline. [42]
Anti-Tau Therapies
Semorinemab:
A tau-targeting monoclonal antibody designed to block extracellular tau and prevent it from spreading between neurons. While its first trials were not successful, ongoing studies are adjusting dosages and targeting different disease stages.
Zagotenemab:
Another anti-tau antibody, zagotenemab is designed to clear extracellular tau aggregates and prevent their propagation. Early clinical trials are underway to evaluate its long-term benefits on cognitive function.
Next-Generation BACE1 Inhibitors
Following the failure of first-generation BACE1 inhibitors, new approaches aim to develop more selective compounds with fewer side effects. These include:
LY3202626:
A selective BACE1 inhibitor that showed some efficacy in early trials, but further research is needed to determine its long-term effects.[43]
5. Innovative Approaches: Combining Therapies
As Alzheimer's is a multifaceted disease, there’s increasing interest in combination therapies that target multiple pathways. Some ongoing studies focus on combining cholinesterase inhibitors, NMDA antagonists, and anti-amyloid or anti-tau therapies to tackle the disease from different angles.
Multi-Target Drugs
Anavex 2-73 (blarcamesine):
A sigma-1 receptor agonist and muscarinic receptor modulator that shows neuroprotective effects. Anavex 2-73 is being studied for its ability to reduce amyloid-beta toxicity, modulate synaptic plasticity, and prevent tau hyperphosphorylation. Phase II trials are promising, and phase III studies are underway.
Tricaprilin:
A ketogenic compound designed to improve mitochondrial function and energy production in neurons. Early clinical studies have shown that enhancing metabolic function may improve cognition in patients with mild to moderate AD.
6. The Future of Alzheimer's Disease Research
The future of Alzheimer's disease treatment lies in early intervention and personalized medicine. Some trends and future directions include:
Advances in fluid biomarkers (e.g., blood or cerebrospinal fluid tests for amyloid-beta and tau) and imaging techniques (e.g., PET scans) are helping to identify Alzheimer's at the preclinical stage. Early diagnosis will allow for earlier treatment, improving the chances of success.
Tailoring therapies based on genetic risk factors, such as APOE status or the presence of specific tau mutations, could lead to more effective treatments. Precision medicine in AD could also involve genetic profiling and adjusting drug combinations to suit individual needs.
Along with pharmacological treatments, research is increasingly focusing on lifestyle interventions, such as diet, exercise, and cognitive training, which have been shown to delay the onset or progression of AD in some cases. [44]
1. Amyloid Beta (A?) Plaques
Overview:
Amyloid beta plaques are aggregates of misfolded amyloid beta peptides, which are believed to contribute to neurodegeneration in Alzheimer's disease.
Drug Targets:
Amyloid-beta Monoclonal Antibodies: Aim to reduce amyloid plaque burden.
Examples:
Aducanumab, Lecanemab, Donanemab.
Tables:
Table 1: Amyloid Beta Targeted Drugs

2. Tau Protein
Overview:
Tau protein aggregates into neurofibrillary tangles, which are another hallmark of Alzheimer's disease.
Drug Targets:
Tau Aggregation Inhibitors: Target tau protein to prevent aggregation.
Examples: TPI-287, LMTX. [44]
Tables:
Tau Tangle Formation
Table 2: Tau Targeted Drugs

3. Cholinesterase Inhibitors
Overview:
Cholinesterase inhibitors increase acetylcholine levels, which is typically decreased in Alzheimer's patients.
Drug Targets:
Cholinesterase Inhibitors: Improve cognitive function by preventing the breakdown of acetylcholine.
Examples:
Donepezil, Rivastigmine, Galantamine.
Tables:: Cholinergic Pathways in Alzheimer's Disease
(Figure is illustrative; please refer to academic sources for actual diagrams.)
Table 3: Cholinesterase Inhibitors
4. NMDA Receptor Antagonists
Overview: NMDA receptor antagonists aim to regulate glutamate activity, which is implicated in neurotoxicity in Alzheimer's disease.
Drug Targets:
CONCLUSION:
The development of effective Alzheimer’s therapies continues to be a challenge, as most current treatments offer symptomatic relief without addressing underlying disease progression. Understanding the role of various receptors, enzymes, and neurotransmitters in AD pathology has opened the door to new therapeutic strategies. Future research will focus on combination therapies and personalized medicine approaches to effectively target multiple pathways involved in Alzheimer’s disease.
REFERENCE

C. Rejitha , A. Hari Nandhini, R. Thelshath, Rajaganapathy Kaliyaperumal, S. Latha, Targeting Alzheimer's Disease: A Comprehensive Review of Current Drug Targets, Receptors, Enzymes, and Neurotransmitters, Int. J. of Pharm. Sci., 2024, Vol 2, Issue 9, 1311-1325. https://doi.org/10.5281/zenodo.13850740
 10.5281/zenodo.13850740
10.5281/zenodo.13850740
