
We use cookies to ensure our website works properly and to personalise your experience. Cookies policy
P.R. Pote Patil College of Pharmacy, Amravati Maharashtra 444604.
Pharmacovigilance (PV) plays a crucial role in ensuring the safety of medicines, but it faces several challenges as drug safety monitoring evolves. One key challenge is the increasing volume and complexity of data, especially with the rise of biologics, personalized medicines, and digital health technologies. Integrating big data, real-world evidence, and artificial intelligence (AI) for signal detection and risk assessment offers significant opportunities for enhancing PV practices. However, data privacy concerns, regulatory hurdles, and the need for standardized data formats remain obstacles. Another challenge is the underreporting of adverse drug reactions (ADRs), which limits the completeness of safety profiles. The expanding use of social media and patient-reported outcomes provides new opportunities to improve ADR reporting, but it also raises issues related to data validation and signal reliability. In the future, PV systems must adapt to emerging therapeutic areas, such as gene therapies and cell-based medicines, which present unique safety monitoring challenges. Regulatory agencies must refine global collaboration and harmonize pharmacovigilance standards to ensure effective monitoring across borders. Advancements in technology, better stakeholder engagement, and improved data integration hold promise to address these challenges and enhance drug safety monitoring worldwide.
Pharmacovigilance, as defined by the European Commission, encompasses the science and activities dedicated to monitoring drug safety. Its core purpose is to minimize risks associated with medication use while maximizing their benefits, ultimately improving patient safety and quality of life. This involves a range of activities, including the collection and analysis of drug safety data, identification of potential new adverse reactions ("signals") through individual case reports, proactive risk management strategies, and effective communication with healthcare professionals, patients, and other stakeholders. This ongoing post-market surveillance is crucial for public health protection, enabling regulatory authorities to update the Summary of Product Characteristics (SPC) for medications based on emerging safety information.
History
The narrative of pharmacovigilance began in 1848 with the tragic death of a young girl, Hannah Greener, following chloroform anesthesia. Despite the widespread adoption of chloroform as a safer anesthetic by Sir James Simpson, the cause of Hannah's death remained a mystery, potentially linked to a fatal arrhythmia or pulmonary aspiration. This incident, along with other anesthesia-related deaths, prompted The Lancet to establish a commission, urging physicians, including those in the colonies, to report such occurrences. These reports, published in 1893, represent an early attempt at systematic data collection related to drug safety. The early 20th century saw further milestones in drug safety regulation. The US Pure Food and Drug Act of 1906 mandated drug purity and prohibited adulteration. This was strengthened in 1911 with a ban on misleading therapeutic claims. A significant catalyst for change was the 1937 sulfanilamide elixir tragedy, resulting in over 100 deaths due to the toxic solvent diethylene glycol. This event led to the 1938 Federal Food, Drug, and Cosmetic Act, a landmark legislation requiring pre-market safety testing for drugs and introducing factory inspections. Also in 1938, the potential link between aspirin and melena was suggested, although its gastrointestinal toxicity wasn't definitively established until 1955, leading to its contraindication in patients with ulcers.A turning point in pharmacovigilance history arrived with the thalidomide disaster in the 1960s. Independent observations by Dr. McBride in Australia and Dr. Lenz in Germany linked thalidomide use during pregnancy to a dramatic increase in congenital malformations. A 1973 retrospective study confirmed this connection. Notably, the thalidomide tragedy was largely averted in the US due to Dr. Kelsey's cautious approach to thalidomide's safety during pregnancy. This catastrophe exposed critical gaps in drug safety practices, particularly concerning animal testing, industry responsibility, and the need for post-marketing surveillance. Crucially, it spurred the evolution of pharmacovigilance from ad-hoc reporting to structured, regulated systems of spontaneous adverse drug reaction reporting, marking a significant shift towards proactive drug safety monitoring.Following the thalidomide tragedy, the landscape of pharmacovigilance underwent significant changes. In 1964, the UK introduced the "Yellow Card" system, a standardized form for reporting suspected adverse drug reactions.

Figure 01: Malfunctioning due to maternal ingestion of thalidomide43.
Around the same time, the US strengthened its drug approval process with a 1962 amendment requiring both safety and efficacy data, including teratogenicity studies in multiple animal species, before a drug could be marketed. Europe followed suit in 1965, developing its own legislation in response to the thalidomide crisis.The mid-1960s also saw the emergence of collaborative research efforts. In 1966, the Boston Collaborative Drug Surveillance Program pioneered epidemiological studies to quantify adverse drug effects through in-hospital monitoring, playing a vital role in shaping the field of drug epidemiology. The World Health Organization (WHO) established its Programme for International Drug Monitoring in 1968, initially with ten member countries, including major players like the UK, US, and Germany. Italy joined this program in 1975. Numerous studies on adverse drug reactions were conducted between 1968 and 1982.The focus on international collaboration continued into the 1990s. In 1992, the European Society of Pharmacovigilance (ESoP) was established, later becoming the International Society of Pharmacovigilance (IsoP), dedicated to advancing all facets of safe medication use. The European Medicines Agency (EMA) was founded in 1995, further solidifying the regulatory framework. In 2001, EudraVigilance, the European database for suspected adverse drug reactions to authorized medicines and those under investigation in clinical trials, was launched. A major overhaul of European pharmacovigilance legislation occurred in 2012 with Directive 2010/84/EU, marking another significant step in the ongoing effort to ensure drug safety.
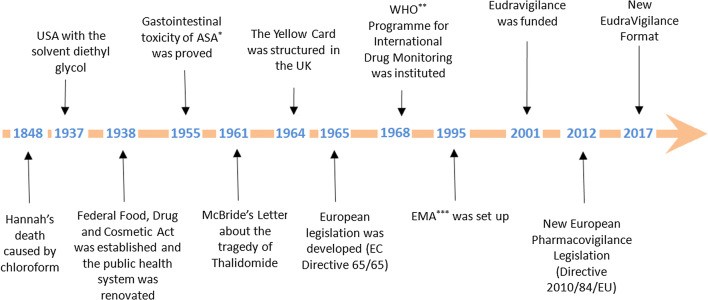
Figure 02: Sequential History of Pharmacovigilance20
How Pharmacovigilance Work in Ensuring Patient Safety in Clinical Studies:
Step 1: Data Collection
Adverse event (AE) reporting is a critical component of pharmacovigilance, enabling the detection and prevention of harmful reactions to medicinal products. Healthcare professionals, patients, and pharmaceutical companies report adverse events to national or international databases, such as the World Health Organization's (WHO) global database, the US Food and Drug Administration's (FDA) Adverse Event Reporting System (FAERS) and the European Medicines Agency's (EMA) Eudra Vigilance. Spontaneous reporting is a voluntary process where healthcare professionals and patients report adverse events, providing valuable insights into medicinal product safety. This approach, however, may result in underreporting, as not all events are captured.
Step 2: Data Processing
Data processing is a critical step in pharmacovigilance, ensuring the accuracy and consistency of adverse event (AE) reports. Data entry involves capturing AE reports into databases, such as the US Food and Drug Administration's (FDA) Adverse Event Reporting System (FAERS) and the European Medicines Agency's (EMA) EudraVigilance. These databases serve as central repositories for AE data, facilitating analysis and signal detection. Following data entry, verification of AE report accuracy and completeness is performed through data validation. This quality control measure ensures the reliability and consistency of the data, preventing errors and inconsistencies that could compromise safety assessments.
Step 3: Signal Detection
Signal detection in pharmacovigilance involves a multi-step process to identify potential safety concerns. Statistical analysis is the first step, utilizing various methods to identify potential safety signals within adverse event data. These statistical methods enable the identification of potential safety signals, which are then further investigated. Building on statistical analysis, data mining is conducted to analyze large datasets and identify patterns. Once potential safety signals are detected, signal validation verifies their accuracy through manual review, causality assessment, literature review, and external data validation. Expert evaluation of adverse event reports determines causal relationships between medicinal products and adverse events, while literature reviews examine scientific literature for supporting evidence. External data validation compares detected signals with external datasets, ensuring reliability. Signal validation informs regulatory decisions, enhances patient safety, and improves medicinal product development. Effective signal detection enables timely identification of safety concerns, informed regulatory decisions, and enhanced patient safety.
Step 4: Signal Evaluation
Causality, severity, and risk-benefit assessments are crucial steps in pharmacovigilance, enabling informed decision-making. Causality assessment evaluates the relationship between a medicinal product and an adverse event (AE). This involves determining whether the AE is directly related to the product, a coincidental occurrence, or exacerbated by underlying conditions. Standardized tools, such as the World Health Organization's (WHO) causality assessment criteria and the Naranjo algorithm, guide evaluators. Causality assessment informs signal validation, regulatory actions, and patient safety measures. Severity assessment evaluates the intensity and impact of adverse events. Severity assessment facilitates the prioritization of safety concerns, regulatory decision-making, and risk management strategies. Risk-benefit assessment weighs the benefits of a medicinal product against its risks. Regulatory agencies, like the FDA and EMA, conduct risk-benefit assessments during product approval and post-marketing surveillance. This assessment ensures medicinal products provide overall benefits, minimizing harm.
Step 5: Risk Management
Risk management is a critical pharmacovigilance step, ensuring medicinal products' benefits outweigh their risks. This process involves three key activities. Risk mitigation implements measures to minimize identified risks, protecting patients from harm. Strategies include dosing adjustments, contraindications, warnings, precautions, and monitoring requirements. Regulatory agencies, manufacturers, and healthcare professionals collaborate to develop and implement effective risk mitigation plans. These plans are regularly reviewed and updated to ensure their effectiveness.This includes updating the product's prescribing information, patient information leaflets, and packaging. Label updates facilitate informed decision-making, enhancing patient safety.
Step 6: Regulatory Agency Interaction
Regulatory interactions are crucial in pharmacovigilance, ensuring transparency, collaboration and compliance. Reporting of adverse event (AE) reports to regulatory agencies, such as the FDA, EMA and WHO, is a critical step. This involves submitting individual case safety reports (ICSRs), periodic safety update reports (PSURs) and risk management plans. Timely reporting enables regulatory agencies to monitor medicinal product safety, identify potential risks and take prompt action. Collaboration with regulatory agencies on safety issues facilitates open communication, ensuring that concerns are addressed proactively. Regular interaction between industry stakeholders, regulatory agencies and healthcare professionals enhances patient safety. Compliance with regulatory requirements is essential, ensuring adherence to established pharmacovigilance standards. Manufacturers must comply with regulations, guidelines and international standards, such as ICH E2A-E2C and GVP modules.
Step 7: Continuous Monitoring
Continuous pharmacovigilance monitoring ensures the safety of medicinal products throughout their lifecycle. Ongoing surveillance involves continuously monitoring adverse event (AE) reports, and identifying trends and potential safety signals. This proactive approach enables timely intervention, minimizing harm to patients. Surveillance activities include: Monitoring AE reports from various sources, such as spontaneous reports, clinical trials, and literature reviews. Summarize safety data from clinical trials and post-marketing experience. Evaluate risks and benefits. Update risk management plans. Signal detection and evaluation is an ongoing process, identifying potential safety concerns through: Statistical analysis of AE reports, Data mining techniques, and Literature reviews.
Step 8: Patient Safety Initiatives
Education and reporting initiatives are vital components of pharmacovigilance, promoting safe medicinal product use. Patient education empowers individuals to manage their health effectively. Clear safety information is provided through patient information leaflets, labelling and healthcare professional counselling. This encompasses: Medicinal product benefits and risks, Proper dosing and administration, Potential side effects and interactions, Importance of adherence and monitoring. Regular workshops, conferences and online resources support ongoing education. Medication error reporting encourages transparency, facilitating prompt corrective action. Healthcare professionals and patients are encouraged to report errors, near misses, a and adverse events 21.
Pharmacovigilance actions:
1. Pharmacovigilance Action
Signal Detection
Reviewing adverse event reports from FAERS and Merck's internal database
Analysing clinical trial data from VIGOR and Approve studies
Evaluating observational studies and literature reviews
2.Pharmacovigilance Action
Data Analysis
Increased risk of myocardial infarction (45% increase)
Increased risk of stroke (50% increase)
Dose-dependent relationship between Vioxx and cardiovascular risk
3.Pharmacovigilance Action :
Risk Assessment
Vioxx increased cardiovascular risk
The risk was dose-dependent
The risk was significant enough to warrant regulatory action
4.Pharmacovigilance Action
Regulatory Action
Reduced prescriptions (95?crease)
Decreased cardiovascular events22.
Good Pharmacovigilance Practices (GVP):
Good Pharmacovigilance Practices (GVP) are guidelines that ensure patient safety by monitoring and managing risks associated with medicinal products. These practices are crucial for pharmaceutical companies, regulatory agencies, and healthcare professionals to minimize adverse reactions and maximize benefits.
GVP Modules:
1. Module I: Pharmacovigilance Systems: Establishing pharmacovigilance systems.
2. Module II: Pharmacovigilance System Master File: Maintaining pharmacovigilance records.
3. Module III: Pharmacovigilance Inspections: Conducting inspections.
4. Module IV: Pharmacovigilance Audits: Performing audits.[23]
5. Module V: Risk Management: Implementing risk management plans.
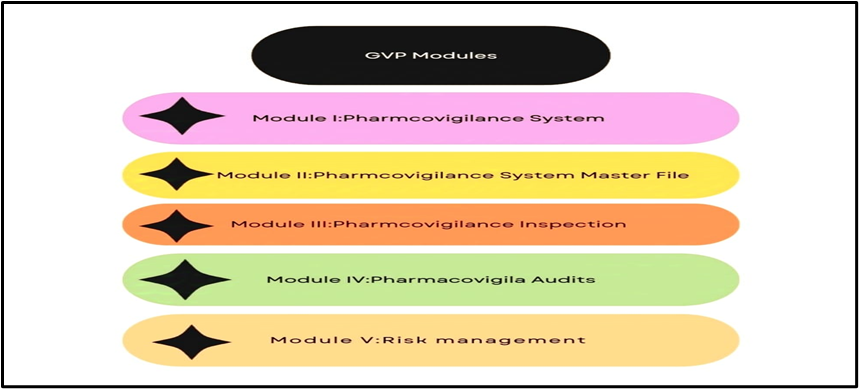
Figure 03: Modules under GVP for Patient Safety
Software Used in Pharmacovigilance:
Let’s look at some software used in Pharmacovigilance for managing and reporting Adverse events.
Table No.01: Lists Of the Software Used in Pharmacovigilance
|
Sr. No |
Software |
|
1. |
Oracle Argus Safety |
|
2. |
ArisG |
|
3. |
Oracle Adverse Event Reporting System (AERS) |
|
4. |
Clin Trac |
|
5. |
Pv NET |
|
6. |
Rep Clinical |
|
7. |
Vigilanz Dynamic Monitoring System |
|
8. |
Web VDME Pharmacovigilance Signal detection and Signal management software |
|
9. |
PV works |
Out of the above software most important two are mentioned below;
1. Oracle Argus Safety
The life sciences industry is increasingly adopting a comprehensive approach to product safety, spanning from clinical trials to post-market monitoring. Oracle Argus Safety is a purpose-built platform designed to meet the intricate pharmacovigilance needs of this sector. This comprehensive solution assists life science organizations in maintaining global regulatory compliance, adhering to standards set by bodies like the EMEA, FDA, and ICH. Argus Safety offers adaptable case management, powerful reporting capabilities, and automation features such as "Auto/Force distribute" and "Auto-Submit," which streamline compliance and lower reporting expenses. The system also supports customizable workflows, wordlist management, and user-defined reporting rules, promoting efficient safety data handling and rapid data entry.Beyond regulatory compliance, Argus Safety tackles the challenges posed by growing case volumes, diverse data sources, and intricate collaborations by integrating safety and risk management functionalities. The platform provides tools for expedited and periodic reporting, adverse event management, and automated risk management, enabling companies to effectively manage a product's benefit-risk profile. Furthermore, Argus Safety integrates with signal detection systems, facilitating proactive safety data analysis and ensuring a holistic safety approach throughout a product's lifecycle, from clinical development to post-marketing. Widely used by leading pharmaceutical, biotech, CRO, and medical device companies, Argus Safety is continually updated and improved through a structured product roadmap, aimed at simplifying processes, minimizing redundant data entry, and boosting productivity23.
2. rep Clinical
RepClinical offers a secure, web-based platform designed to streamline essential pharmacovigilance tasks efficiently and cost-effectively. It enables users to capture adverse event data, produce regulatory reports, and exchange Individual Case Safety Reports (ICSRs) with various regulatory agencies and business partners, all within a user-friendly and efficient environment. The system features uncluttered screens and helpful tools for generating accurate E2B reports with ease.RepClinical's data structure closely mirrors that of ICSRs. Case data is stored and tracked within "Cases," while administrative and identification details are managed within "Safety Reports." Message data is stored and tracked separately in "Messages." Users can create, modify, and monitor cases, assigning them to others for updates or review. One-click access to case data simplifies retrieval without complex menu navigation. RepClinical also incorporates early-stage data checks during entry to ensure data quality.Safety reports, linked to individual cases, can also be created, modified, and tracked within the system. A safety report can only be generated if a corresponding case exists. Like cases, safety reports can be assigned to other users for review and modification. Creating messages in RepClinical is a quick and straightforward process. These messages can be archived for later retrieval and are automatically validated by the system24.
Future Prospective of Pharmacovigilance –
Pharmacovigilance has evolved significantly since its formal recognition in the 1960s, spurred by tragedies like the thalidomide crisis. More recent incidents, such as the aprotinin withdrawal and concerns surrounding rosiglitazone's safety, underscore the public's deep interest in drug safety. Current pharmacovigilance systems are undergoing significant revisions to address future challenges. Establishing a strong scientific foundation is crucial for the field's advancement and its contribution to innovation. Future pharmacovigilance practices must rapidly detect emerging safety concerns. Success in this area will likely bolster public trust in medications. Additionally, pharmacovigilance methodologies should be able to pinpoint patient populations susceptible to adverse drug reactions (ADRs) and characterize the progression of these reactions. Increased patient involvement as a source of information represents a promising avenue for achieving these goals, aligning with the broader trend of greater patient participation in drug safety.Artificial intelligence (AI), particularly machine learning techniques like natural language processing and deep learning, offers the potential to automate and enhance pharmacovigilance by identifying and extracting information about adverse drug events. Furthermore, the growing reliance on telehealth for managing various health conditions creates opportunities for AI to contribute to the detection and prevention of such events. This review explores two specific examples of how AI can improve pharmacovigilance quality and its application within telehealth settings.25, 26
CONCLUSION
From this review, it was concluded that pharmacovigilance becomes an effective tool in ensuring drug safety, protecting public health, and maintaining trust in healthcare systems. Pharmacovigilance plays an important role in clinical trials in ensuring patient safety through data collection, data processing, signal detection, signal evaluation, risk management, regulatory agency interaction, continuous monitoring, and patient safety initiatives. Pharmacovigilance shows its action by signal detection, data analysis, risk assessment, and regulatory action it also includes GVP i.e. good pharmacovigilance guidelines. GVP has several modules such as Module 1, Module 2, Module 3, Module 4, and Module 5. Pharmacovigilance also includes several software such as Oracle Argus Safety, ArisG, Oracle Adverse Event Reporting System (AERS), ClinTrac, PvNET, repClinical, Vigilanz Dynamic Monitoring System, WebVDME, Pharmacovigilance Signal detection and Signal management software, PV works. For the future perspective of pharmacovigilance, modern pharmacovigilance needs to quickly identify new drug safety issues to maintain and improve public confidence in medications. Effective methods should also be able to identify patient groups at higher risk for adverse drug reactions (ADRs) and understand how these reactions develop. Encouraging patients to actively contribute information is a promising strategy for reaching these objectives and reflects the growing trend of patient engagement in drug safety.Artificial intelligence, especially machine learning approaches such as natural language processing and deep learning, can revolutionize pharmacovigilance by automating the identification and extraction of adverse drug event data. The increasing use of telehealth also provides new avenues for AI to improve the detection and prevention of these events. This review examines two concrete examples of how AI can enhance pharmacovigilance, including its application in telehealth.
CONFLICT OF INTEREST: Authors have no conflict of interest for regarding this investigation.
ACKNOWLEDGEMENT: Authors are thankful to P. R. Pote Patil College of Pharmacy, Amravati for their valuable support and guidance to carry out this work.
REFERENCES


Abhijeet Welankiwar*, Anchal Kulte, Pradyumna Keche, The Impact of Pharmacovigilance on Patient Safety: A Comprehensive Review, Int. J. of Pharm. Sci., 2025, Vol 3, Issue 2, 1220-1228. https://doi.org/10.5281/zenodo.14875777
 10.5281/zenodo.14875777
10.5281/zenodo.14875777