
We use cookies to ensure our website works properly and to personalise your experience. Cookies policy
Dr. Shivajirao Kadam College of Pharmacy, Kasabe Digraj, Sangli (MS), India. 416305.
The primary cause of dementia, Alzheimer's disease (AD) is a progressive neurological illness marked by behavioral abnormalities, memory loss, and cognitive decline. Amyloid-beta (A?) plaques and hyper phosphorylated tau tangles are the main pathophysiological components of AD, resulting in synaptic dysfunction, oxidative stress, and neuronal death. While the specific cause remains unclear, various ideas have been presented, including the amyloid cascade, tau pathology, oxidative stress, cholinergic dysfunction, glutamate excitotoxicity, inflammation, and mitochondrial dysfunction. The start and course of illness are also influenced by environmental and genetic risk factors, including the APOE ?4 allele, age, food, physical inactivity, smoking, diabetes, and hypertension. Together with neuroimaging methods, biomarkers including A?42, total tau, and phosphorylated tau levels in CSF fluid are essential for early diagnosis and follow-up. Cholinesterase inhibitors and NMDA receptor antagonists, such as donepezil and memantine, are the mainstays of current therapy, which alleviate symptoms. New treatment strategies focus on neuroinflammation, tau aggregation, A? generation, and synaptic repair. A proven cure or extremely successful disease-modifying medication is still elusive after decades of study. There is promise for early intervention and disease control because to ongoing clinical studies and biomarker developments.
The most prevalent type of cognition in senior citizens, Amyloid-β plaques and tau tangles are hallmarks of Alzheimer's disease (AD), which results in neuronal death early detection and identification of at-risk persons is critical for prevention. Biomarkers involve Aβ42 as well as entire the protein tau concentrations within cerebrospinal fluid and phosphorylated tau, as well as brain imaging for amyloid deposits, the process of atrophy and the metabolism of glucose. [1]. The majority of Alzheimer's disease (AD) cases are sporadic, with less than 10% resulting from changes in the genes for APP, presenilin 1, and presenilin 2 that produce Aβ. People with Down syndrome are at risk syndrome frequently acquire Alzheimer's-like dementia as a result of APP overexpression. The specific role of Aβ and tau in Alzheimer's disease onset is unknown, although being essential to study. In 2006, 26.6 million people worldwide had Alzheimer's disease, with forecasts of 106.8 million by 2050. [2]. the development of tangles composed of tau and amyloid-beta plaques in Alzheimer's disease mostly harms the neocortex and hippocampal regions, which are important sites for memory. This leads to synapse disruption and neuronal death, reducing memory recall and meaningful communication. [8]. In recent years, two identical hypotheses were developed categorize the three stages of Alzheimer's disease progression. The following three phases of Alzheimer's disease were proposed by the International Working Group for Alzheimer's Disease (IWG): (1) asymptomatic at-risk (no symptoms and AD pathology demonstrated by biomarkers), (2) premature Alzheimer's disease (episodic memory deficit with damaged cued recall that may occur alone or in conjunction of additional cognitive changes and AD biomarker evidence), and (3) AD dementia.[3] The task force examined previous unsuccessful studies and underlined the importance of biomarkers and novel trial designs in improving efficiency and detecting therapeutic results earlier. Concerns are growing that treating It might be too late treat low moderately severe AD to improve results. This annual update assesses current phase 1–3 clinical trials and spotlights continuing novel therapeutics for Alzheimer's disease.[4] ] Expanding on previous contributions, we In our yearly report on the status of the medication development pipeline, we assess all of the phase one phase two, and phase three research studies currently underway in AD. We outline experimental therapies and clinical trials for AD neuropsychiatric symptoms, cognitive improvement, and disease modification. [9]

Figure 1-2019 Alzheimer’s Drug Development
Alzheimer's disease has no known cause, although important risk factors include aging, genetics, and the environment. There is presently no cure. Early discovery is critical, as researchers continue to investigate brain changes to improve diagnosis, prognosis, and prevention. [5] Biomarkers, both chemical and imaging, reveal particular alterations in Alzheimer's disease that occur in a distinct order. Longitudinal studies now routinely test these indicators in order to track their evolution and correlate them with clinical complaints. [6]. Alzheimer's research has switched from therapy to prevention, with the goal of stopping both disease start and symptom development. However, significant hurdles persist in coordinating efforts to identify effective preventive methods. [4]
Alzheimer's disease risk rises with age, from approximately 3% at age 65 to more than 30% by age 85, impacted by environmental and genetic factors. [6] Alzheimer's disease is classified as either late-onset (LOAD, over age 65) or early-onset illnesses (EOAD, before age 65) with EOAD accounting for 1-5% of cases. EOAD frequently exhibits a strong familial pattern and Mendelian inheritance, whereas LOAD is more random. Heritability can reach 79%, with EOAD and LOAD having different genetic profiles. [7]
Alzheimer's disease with an early onset
Alzimers with Early Onsets Disease [EOAD] is often caused by rare, widely distributed changes in three genes that encode proteins involved in the breakdown of amyloid precursor proteins (APP) and the synthesis of Aβ: APP, presenilin 1 (PSEN1), and presenilin 2 (PSEN2).[7]
Alzheimer's disease that develops late
Risk factors for Alzheimer's disease that develops later (LOAD) include genetic, behavioural, and environmental variables. The allele of ε4 in the APOE Chromosome 19q13.2 encodes the gene most powerful known risk factor for heredity. However, other non-genetic factors also have a significant impact on how illnesses develop. [7] Alzheimer's International convened specialists in 2005 to determine dementia prevalence, which revealed 24.2 million sufferers and 4.6 million new cases per year worldwide. Identifying the onset age and designating unaffected populations make it challenging to determine the exact AD incidence. As people age, incidence rates increase, starting at about 0.5% annually from ages 65 to 70 to 6-8% at age 85. The significant global prevalence reflects AD's extended duration and increasing frequency with age. Advances in standardized diagnostic technologies now enable more accurate comparisons of Alzheimer's disease rates across communities. [10] Given the strong correlation between AD development and aging, the dementing disease is predicted to pose significant challenges to public health and elder care systems across the world.[11] People with dementia frequently have a much reduced quality of life, owing to economic difficulties and social isolation. The stigma associated with the disease often drives these issues. [12]
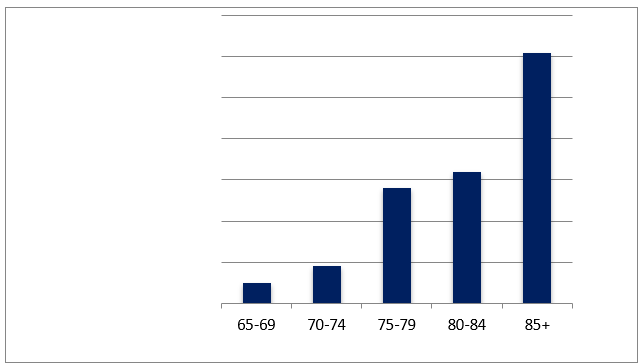
Figure 2. The annual prevalence rate of Alzheimer's disease
Pathophysiology Of a Disease of Alzheimer
Important brain regions like the hippocampus experience neuronal loss and degradation in Alzheimer's disease. It is distinguished via loss of neurons, inflammation, tau tangles and amyloid plaques. This leads to increased monocyte and macrophage infiltration and microglial activation in the brain. [13]
Hyper phosphorylated the amyloid beta hypothesis and tau protein
The processing of an amyloid precursor protein (APP) occurs in peripheral tissues by two pathways: non-amyloidogenic, where α-secretase cleavage prevents Aβ generation, and amyloidogenic, where β-secretase cleavage results in β-amyloid peptide formation after γ-secretase action. These pathways determine if dangerous Aβ peptides are formed. [14].
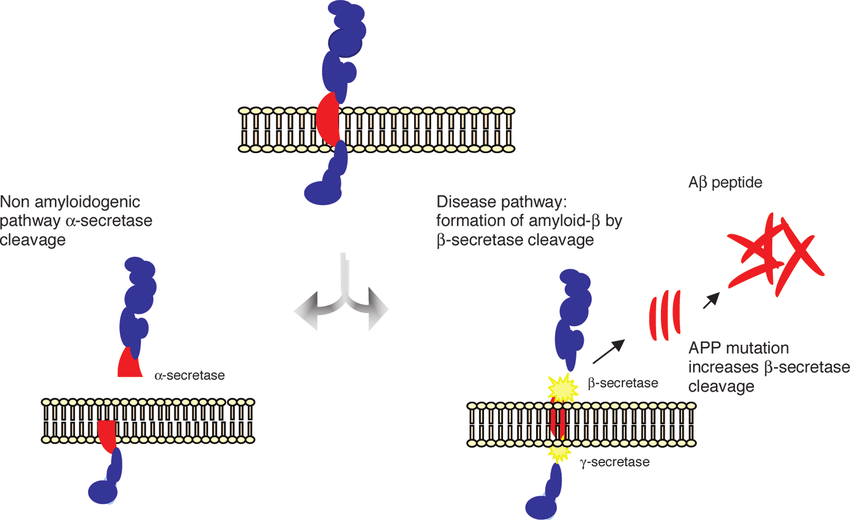
Figure 4: APP Cleavage Pathways in Alzheimer's
Secretases break the APP, or amyloid precursor protein by two pathways: nonamyloidogenic (α-secretase inhibits deleterious peptide synthesis) and amyloidogenic Neurotoxic Aβ peptides are produced by β- and γ-secretases. Asymmetry in Aβ synthesis and clearing leads to hazardous oligomers and plaques. Aβ toxicity and aggregation are influenced by factors such as its sequence, concentration, and stability. [13]
Oxidative stress theory
Reactive oxygen species (ROS) and reactive nitrogen varieties (RNS) are produced by a variety of normal and pathological activities. By damaging cell structures, these chemicals can have both beneficial and negative effects on cellular signaling. Due to its high oxygen consumption—roughly 20% more than that of other mitochondrial tissues—the brain is especially susceptible to oxidative injury.Brain's neurons core units, include significant quantities of Fatty acids with polyunsaturated chains, which easily interact with ROS, resulting in lipid peroxidation and cell death. Furthermore, neurons have low glutathione levels, which limits their ability to withstand oxidative damage. This combination causes substantial oxidative stress harm in the brain. [15]
Metal Ion Hypothesis
The Alzheimer's disease metal hypothesis associates aberrant metal levels with neurofibrillary tangles and amyloid plaques. Chelators that bind copper and zinc, such as clioquinol, can help restore metalloprotease function and have demonstrated encouraging benefits in AD mouse models. [16] Copper (Cu) and zinc (Zn)-containing extracellular amyloid plaques are a hallmark of Alzheimer's disease, with axonal Cu deficit contributing to pathogenesis. Metal-chelating medicines can dissolve plaques by disrupting the interactions between Zn and Cu ions, which influence Aβ formation and stability. The metal- 5-chloro-7-iodo-quinolin-8-ol (clioquinol, CQ) is a chelator. Reduces Aβ toxicity by increasing Zn and Cu absorption and triggering protective cell signaling that breaks down Aβ. In a phase II experiment, CQ decreased cognitive deterioration in those with Alzheimer's and was well tolerated. [17] Observed that AD patients had the same quantity of copper in their plasma as gender-matched and healthy age controls. Abnormal copper levels can harm the nervous system and raise the likelihood of developing Alzheimer's disease. [18]
Cholinergic hypothesis
The neurological condition known as Alzheimer's disease, or dementia, affects the hippocampus and neocortex, two areas of the brain that gradually deteriorate. Alzheimer's disease is characterized by neurofibrillary tangles, β-amyloid plaques, and the death of neurons and synapses.Figure 3 depicts how these alterations cause the disease's usual symptoms. [19] Although there is debate regarding which transmitter systems undergo the most significant alterations alongside ordinary aging and whether this sequence varies in brains affected by Alzheimer's disease, the hypothesis of cholinergic dysfunction focuses on identifying functionally important modifications in the essential cholinergic nerve system within aged brain tissue. [20]
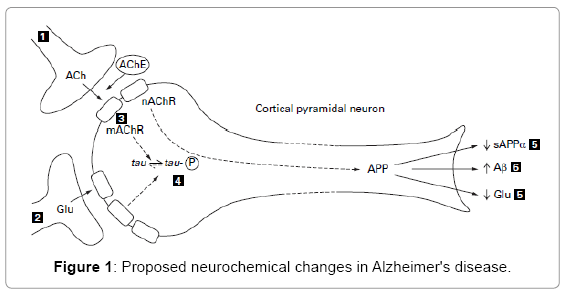
Figure -5 The Neurochemical change in Alzheimer’s disease.
Concept of glutamatergic/excitotoxicity
Cholinergic neurotransmitters in the central nervous system are connected to glutamatergic and neurons, which are critical for cognition.Alzheimer's disease pathogenesis includes alterations in glutamate receptor levels, particularly the NMDA receptor. Alzheimer's disease is characterized by persistently low levels of NMDA receptor stimulation, which results in reduced neurotransmission. [21] Abnormal NMDA receptor modulation results in prolonged calcium influx, which causes neuronal damage and signal disruption. Increased APP synthesis increases plaque formation and tau hyperphosphorylation, which results in neurofibrillary tangles and toxicity. Glutamate is removed by reuptake, and tau hyperphosphorylation is thought to occur after plaque development, however the exact source is unknown. [21]
Tau Hypothesis
Tau, a microtubule-associated protein, helps axons, but aggregation causes neuronal injury and degeneration. Targeting tau, which is intimately connected to AD development, has been a primary focus for treatment after Aβ treatments failed. [22] Hyperphosphorylation and aggregation of tau, regardless of mutations, lead to active disease during aging. Inhibiting this mechanism could potentially prevent, hold up, or even reverse Alzheimer's disease. [23] Phosphorylation, a amino acid called monomethylation, lysine in acetylation, lysine in monomethylation, lysine in dimethylation, lysine in ubiquitylation, and serine are among the modifications that tau experiences. [22]
Inflammation Hypothesis
Epidemiological study supports the inflammation hypothesis by showing that long-term anti-inflammatory therapy in rheumatic arthritis patients is linked to a lesser incidence of Alzheimer's disease. Anti-inflammatory medications may help delay the onset of Alzheimer's disease, according to observational studies. Additionally, nitric oxide-releasing NSAIDs have been shown in human studies and animal models to reduce or stop the course of Alzheimer's disease. [23] Recent discoveries about microglial dysfunction in neurogenesis and plasticity provide new avenues for Alzheimer's diagnosis and treatment. Restoring microglial balance may provide new therapeutics, however specific biomarkers indicating microglial function are urgently required. This supports the idea that Alzheimer's disease is inflammatory, as shown in Figure 6. [23]
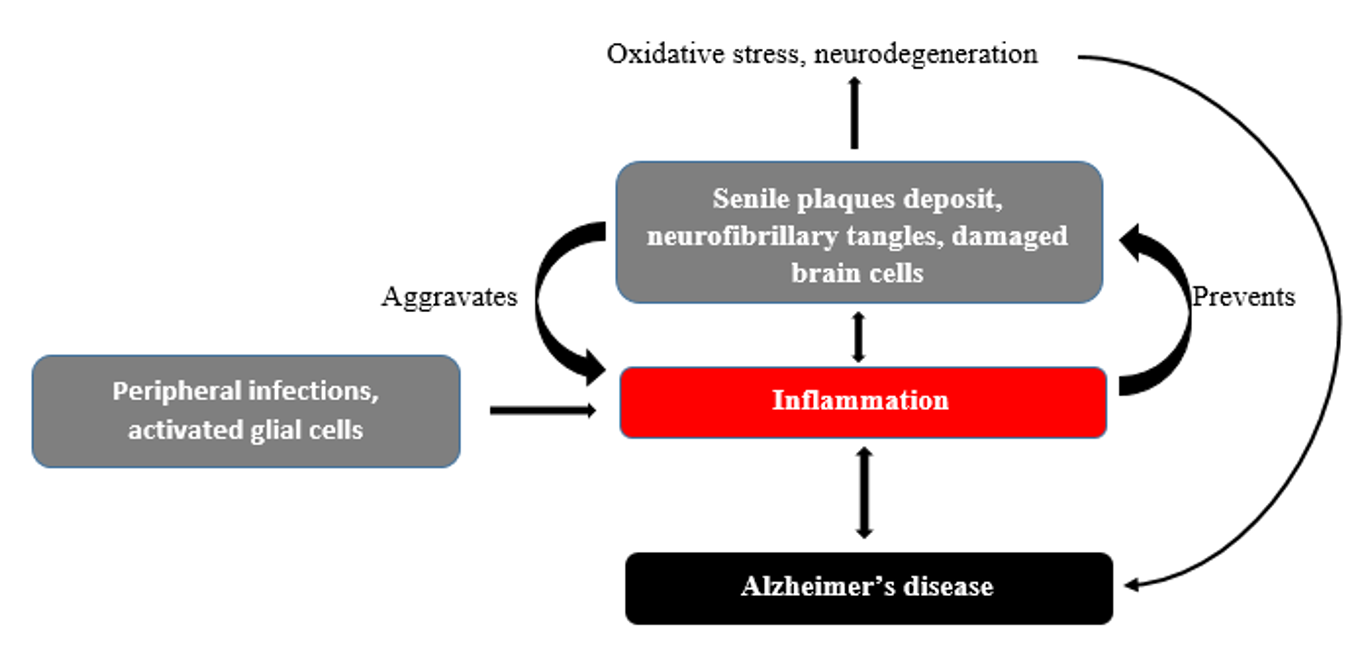
Figure 6-Inflammatory Hypothesis.
The Amyloid Cascade Theory
The Amyloid Cascade Alzheimer's disease is thought to be brought on by the buildup for amyloid particles fi protein (A3P) that is the main component of plaques. This deposition directly causes dementia, vascular damage, neurofibrillary tangles, and cell death. The peptide product known as APP is produced by the larger amyloid precursor protein (APP). [24] The fundamental tenet of the amyloid hypothesis, first proposed in the first decade of the 1990s (Hardy & Higgins, 1992; Selkoe, the year 1991), argues that either increased or reduced removal of Aβ peptides causes the disease. Hydrophobic material Aβ40 and Aβ42 peptides accumulate and combine resulting in insoluble plaques, which trigger a cascade of detrimental events that ultimately result in AD and neuronal death. The primary shift in this idea over time has been the explanation of the pathogen Aβ that is believed to initiate detrimental processes and cause AD. [25]
Mitochondrial Cascade Hypothesis
Our own studies on the causes and outcomes of AD-related mitochondrial dysfunction have had a significant influence on our mitochondrial cascade hypothesis, which we initially put forth in 2004. [26]. we must emphasize that finding dysfunctional mitochondria in AD patients was by no means our first discovery. The altered mitochondrial shape in AD sufferers' brains has long been known.Deficits in the pyruvate dehydrogenase complex and ketoglutarate, two Krebs cycle enzymes. [27]
- Stroke
- Atrial Fibrillation
- Heart Failure
1] A Factor of Genetic Risk
The age at which the initial symptoms appear can be used to define AD. Approximately 4–6% of AD cases are early-onset, affecting persons under 65, while late-onset dementia affects people 65 and older. The early and late forms of AD differ in clinically intellectual, neuropathological, and neuroimaging aspects, as well as in the age at which symptoms first manifest. [28] Found that over 70% of the risk of developing Alzheimer's disease is influenced by heredity. The genes that make up APP, PSEN1, and PSEN2 are usually mutated in early Alzheimer's disease.[28] In 1990, Levy et al. found that patients with hereditary cerebral hemorrhage with Dutch-type amyloidosis carried a Glu693Gln mutation in missense (app770 codon numbering). Many missense variations in the APP gene's exons 16 and 17 have been found in individuals with early onset in recent years. [29]. APP mutations were found to be an uncommon cause of Alzheimer's disease, affecting just 5% of families with early onset. Mutations in the PS1 and PS2 genes are responsible for around 50% of FAD cases. Since 1995, 80 individuals with early-onset FAD from a variety of ethnic backgrounds have been found to have over 50 PS1 mutations.PS1's coding area consists of ten exons (numbered 3 to 12). [29] The physiology of apolipoprotein E and its role as a familial risk indicator for Alzheimer's disease will be the main topics of discussion. Only one amino acid separates the three isoforms of F apolipoprotein E, which is a 35 kDa protein (E2, E3, and E4). The ε2, ε3, and ε4 alleles that code for the f of apolipoprotein E isoforms are approximately 7–8%, 75–80%, and 14–15% prevalent in the general population, respectively. [30]
2] Diet
For several decades, experts have discovered a link between nutrition and the risk of acquiring Alzheimer's disease (AD). Certain dietary patterns may raise the risk of acquiring Alzheimer's disease, while others may guard against it. The Mediterranean diet has preventive advantages since it is high in fruits, vegetables, aquatic life, unsaturated fats, and antioxidants. A greater chance of Alzheimer's disease has been linked to diets high in saturated fats, Tran's fats, and inadequate antioxidants. Alzheimer's disease has been associated with diabetes, high blood pressure, obesity, and high cholesterol.[31] Numerous nutrients, such as antioxidant substances, choline, and omega-3 fatty acids, have been suggested to affect cerebral functioning since the early 20th century, when it was realized how crucial B-vitamins were for neuronal function and cognition.[31]
3] Smoking
"Smoking" refers to the continuous use of tobacco in the form of tobacco. Tobacco products, primarily cigarettes, are used by around 2 billion people worldwide. Tobacco-related diseases cause at least 4 million deaths globally. [32] While the neuroprotective effects of nicotine and medications containing nicotine are yet unknown, smoking seems to lower the possibility of Alzheimer's disease, according to Newhouse, Potter, and Levin (1997). Smoking appears to reduce the risk of Alzheimer's disease (AD).
Neuroprotective impact of nicotine and nicotine-containing medicines is unknown (Newhouse, Potter, & Levin, 1997). [33]
4] Depression
Fiske et al. (2009) state that early or midcentury depression and declining health and function are two factors that contribute to late-life depression. Chronic illness, functional disability, and a history of mental illness raise the risk of developing depression, according to prospective cohort studies of older persons in the general population without cognitive impairment. While these models may be applicable to individuals with dementia, there may be significant variations depending on the degree and level of insight.
Psychosocial components and other neuropsychiatric problems. [34]Depressive symptoms are common in AD, impacting 20–30% of people. About 300 million people worldwide suffer from depression, a serious medical condition that can worsen pre-existing conditions and increase functional impairment. Clinical research reveals an association between depression and Alzheimer's disease. It is still unclear if depression is a risk factor for Alzheimer's disease, an early sign of neurodegeneration, or a reaction to early cognitive abnormalities. [35]
5] Alcohol Consumption
Excessive alcohol use affects brain structure and neuropsychological functioning, impairing cognitive function both immediately and over time. Because brain injury encourages brain atrophy and shrinkage, which is a key indicator of neurodegenerative changes and cognitive impairment in aging, it is linked to clinical dementia. However, while atrophy decreases and cognitive function recovers after alcohol cessation, these morphological changes brought on by alcohol use may be reversible, unlike AD or aging. [36]
6] Physical Activity
Reduced a substance called generation, increased β-amyloid elimination, improved blood flow and vasculature in the brain, anti-oxidative as well inflammatory responses in the brain's activity, as well as indirect mechanisms through improvements in mood, sleep, and other the cardiovascular risk factors are some of the ways that regular physical activity (PA) can prevent dementia. [37]
7] Cerebrovascular Disease
Since CVD and AD show additive clinical and pathological effects, there is growing evidence that they interact at the cellular level, potentially worsening both. [38] Alzheimer's disease has been linked to cardiovascular diseases (CVDs), which are common in the elderly and include heart failure, stroke, atrial fibrillation, and CHD.[39] Vascular dementia is the second most common cause of dementia, accounting for the vast majority of cases, behind Alzheimer's disease (AD). Individuals with cardiac risk factors are most affected by vascular dementia. Nonetheless, those who are diagnosed with Alzheimer's disease share the same risk factors. [40] Age, previous health history, way of life, inheritance, and clinical factors that modify morphological and hemodynamic function are some of the variables that affect the outcome of cardiovascular disease. The arterial walls and structural tissue of the heart are impacted by cardiovascular disease. It can be categorized as heart failure, arrhythmia, congenital, cardiomyopathic, valvular, or coronary. Brain hypoperfusion can arise from cerebral autoregulation balance being disrupted by coronary artery disease stiffness of the arteries, or cardiovascular disease (Figure 7). [41]
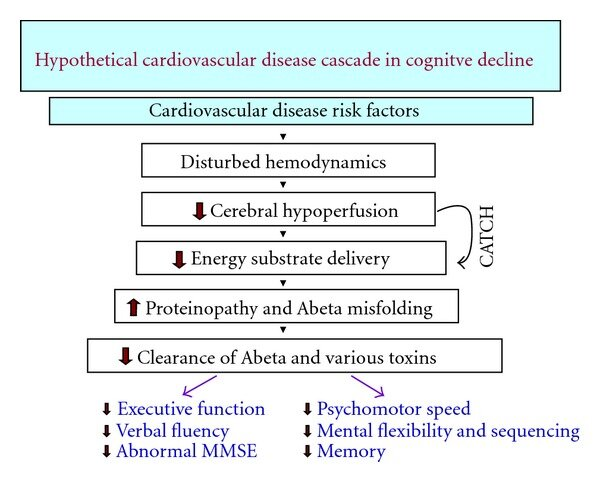
Figure 7 CATCH Hypoperfusion Cascade.
8] Diabetes
The insulin-resistance syndrome includes diabetes, insulin resistance, a high level of impaired glucose tolerance, elevated cholesterol with/or triglyceride concentrations, insufficient good cholesterol levels, elevated pressure, and obesity that are separate indicators of risk for cardiovascular disease (CAD). Type II diabetes has insulin resistance syndrome as a precursor numerous studies have now linked diabetes and insulin-resistant syndrome to AD. [42] Type 2 diabetes (DM) is a long-term metabolic condition that primarily affects the elderly. As a significant geriatric cardiovascular risk indicator, it has been studied in relation to deposits of amyloid in the brain of a person.This neuropathologic approach is primarily necessary in the definition of Alzheimer's disease, according to the new National Institute on Aging and Alzheimer's Association (NIA-AA) research framework (2018), which emphasizes the use of biomarkers. [43] Alzheimer's illness is caused includes amyloid beta (Aβ) proteins lesions that are composed of fibrillar Aβ, neurofibrillary tangled structures (NFTs) composed of increased phosphorylation proteins known as tau, and chronic neuroinflammation, which promotes both diseases. [44] The metabolic syndrome, which encompasses obesity, insulin resistance, coronary artery disease, hypertension, and lipid disorders, increases the possibility of Alzheimer's disease over that of type 2 diabetes. Obesity, type 2 diabetes, and Alzheimer's disease all share similar molecular disorders, like insulin resistance. Oxidative stress, mitochondrial dysfunction, and inflammation. [44]
9] Hypertension
Hypertension in the middle decades has been recognized as an indicator of risk for dementia and Alzheimer's, with multiple research establishing a relationship between the two disorders. Hypertension can be caused by inadequate both diastolic and systolic blood Normal systolic blood pressure for adults is <120 mm Hg, whereas the diastolic reading is <80 mmHg. [45] Cognitive function has a complex association with hypertension progression. The Honolulu Aging study indicated that people with Stage 2 hypertension (BP>160/95) had poorer cognitive impairment compared to those with Stage 1 hypertension (BP>140/90). Although minor, this effect is supported by extensive epidemiological evidence. The impact of stage 2 hypertension may be overestimated compared to stage 1. Individuals with stage 2 hypertension are more likely to die from all causes and have a lower life expectancy. While AD is widespread. [46] Many research have concentrated on the potential mechanisms underlying blood in the cerebral cortex artery walls, which are the brain's initial layer of defense against peripheral problems including hypertension. Tension. Endothelial cells contain the RAGE receptor, which we discovered that the initial hypertensive stimulus increased the enzyme RAGE activation in thinking brain areas. A recent study looked at how the angiotensin-2 II (AngII) first-generation receptor system influences RAGE synthesis in developed endothelial cell lines (ECs) from people with essential hypertension. AngII increased RAGE mRNA levels among micro ECs that are and solubility RAGE synthesis in the EC medium, suggesting that the interaction between renin and angiotensin plays a role in vascular angiopathy development and progression. [47]
10] Cardiovascular disease
CVDs, such as cerebrovascular accident, ventricular fibrillation (AF), cardiac disease (CHD), and cardiac failure, are common in the elderly and often related with Alzheimer's disease. The relationship connecting CVDs along with AD may be due to common danger factors, yet it might not be a direct reason. Cardiac disease has been associated with hypo perfusion and microemboli. [48]
Stroke
Clinical research has demonstrated that the co-occurrence of AD and stroke happens more frequently than would be predicted by chance, suggesting a potential link between the two conditions despite the distinct etiopathogeneses for AD and stroke that have been proposed. The exact association between Alzheimer's disease and stroke is still uncertain. [49] As individuals age, their probability of stroke increase. Additionally, AD gets increasingly prevalent with age; in adults over 75, its prevalence might reach 20%. In addition to common risk factors like diabetes or hypertension, AD may also be a risk factor for stroke, as evidenced by the potential correlation between an AD diagnosis and a subsequent stroke. Determining if Alzheimer's disease is an increased risk cause of stroke is so essential to clinical care and public health. [50]
Atrial Fibrillation
Alzheimer and heart failure (AF) often coexist and mostly affect the elderly. [51] Whether or not AF causes a stroke that is ischemic, it can also produce reduced cerebral perfusion due to decreased cardiac output, therefore may eventually result in vascular dementia, another form of hypoperfusion genesis.In fact, preliminary studies employing rodent models have indicated that cerebral hypoperfusion results in working memory impairment and white matter alterations such demyelination and axonal damage. [52]
Heart Failure
Heart failure and Alzheimer's disease frequently coexist, which raises care costs and the use of medical resources. [53] A number of studies have shown that because HF increases neuro hormonal activity and decreases blood supply to the brain, resulting in neuronal energy crises and malfunction of the neurovascular unit, it may boost the chance of developing AD. The amyloidogenic pathway is initiated by chronic reduced blood flow to the brain caused by HF, which converts APP into neurotoxic A (2). Long-term cerebral hypoperfusion has been shown to increase β-secretase levels in an experiment. Because HF causes prolonged cerebral hypoperfusion, which weakens the blood-brain barrier and causes unregulated glial activation, it worsens cognitive impairment. It's intriguing to notice the breakdown between the heart-brain circuits seems to be an important factor in the emergence of Alzheimer's disease and mental decline in HF patients. [54]
4. Biomarkers For Alzheimer’s Disease
The three primary indicators of dementia (AD), amyloid B (Ab42), total tau (T-tau), as well as phosphorylated tau protein (P-tau), are supported by the accumulation of data from clinical research to reflect essential elements of AD pathogenesis. Crucially, several clinical investigations repeatedly demonstrate that these biomarkers provide diagnostically significant information, even at the earliest stages of disease. [55] Alzheimer's disease (AD) with a focus on uses for diagnostics. The most widely used ELISA techniques, the INNOTEST assays, for measuring total tau (T-tau), phosphorylated tau (P-tau), and amyloid-b (Ab42) in CSF were published more than 20 years ago. These assays demonstrated higher levels of T-tau and P-tau, as well as reduced Ab42, which represents a pattern known to represent the "Alzheimer's CSF profiles."[55]
Amyloid B
The deposition of amyloid beta (Ab) in senile deposits in the cortex of the brain is an early step in the progression of Alzheimer's disease. In the amyloidogenic process for APP route, b- and g-secretases break the protein called amyloid precursor (APP) sequentially to produce Ab's longest isoform, which contains 42 amino acids. The b-secretase activity comes from central barrier aspartyl protease to and this is produced from the b site APP splitting enzyme 1 gene (BACE1). The presenilin is (PS1 and PS2) proteins are the catalysis nucleus on g secretase, an intramembrane splitting complex composed of at least four subunits. [56] Postmortem investigations show an inverse relationship between Ab42 level throughout CSF and the number of amyloid plaques. Additionally, PET tests employing Pitts compound B (PIB) produce a positive signal with approximately 100% concordance. Clinical investigations involving 20,000 alzheimer patients and controls identified a 50% reduction in Ab42 levels in the Cerebrospinal within those with Alzheimer's in comparison to control systems, overall predictive diagnostic accuracy and specificity somewhere between 80% to 90%. Studies on AD mouse models suggest that plaques operate as an "A 42 sink," limiting the transit of soluble A 42 from the brain to the cerebrospinal fluid. [56, 57] Human amyloidosis precursor’s protein in them, a kind 1 transmembrane protein, generates Ab via metabolism. Alzheimer's disease can be inherited autosomally by mutations in the APP or presenilins. These mutations alter APP processing, resulting in increased production either Ab either a 42-amino-acid version with a greater proclivity for plaque formation. This shows that disrupted APP processing is a factor in familial AD3-6. Ab's neurotoxicity indicates that it is directly related to neurodegeneration. In vitro, Ab neurodegeneration is enhanced by peptides aggregation and the formation of amyloid fibrils identical to those encountered in complete amyloid plaques. [58] The enzymes α- and gamma-secretase degrade the precursor protein for amyloid (APP), a membrane-spanning polypeptide having a single transmembrane domain, resulting in Aβ. Presenilins, nicastrin, front pharynx defective-1, and presenilin enhancer-2 all constitute the active components of the gamma-secretase complex, as shown by various studies. Gamma-secretase's molecular structure remains constant across metazoans and serves a purpose. The primary enzyme liable for Notch signalling and membrane receptor processing performs a variety of important activities.[59] The analysis of serum Aβ levels among those with Alzheimer's has been conflicting, with some research showing arise in Aβ1−42 and a decline in Aβ1−40, whereas other studies reported contradictory results. [60] According to the most recent study, APP transgenic mice show spontaneous nonconvulsive convulsions, indicating increased excitation, while Aβ decreases synaptic activity in vitro. Reduced concentrations within the GluR1 and GluR2 AMPA transmitter subunits and modifications in the phosphorylation condition of the NR2B subunit of the NMDA receptor indicated changed glutamate receptor regulation. These results are significant, but particularly in light of new data showing that people with Alzheimer's disease had more seizures. Moreover, the clinical effectiveness of memantine, which an NMDA receptor inhibitor that slows the progression of AD, would be contextualized by harmful overexcitation of cortical networks. In contrast to the overexcitation observed in the APP transgenic mice, electrophysiological research finds that Aβ has an initially suppressive effect on synaptic transmission. [61]
Tau Protein
In the 1970s, while studying elements involved in microtubule formation, tau protein was discovered. A crucial part of the neuronal cytoplasm that supports and shapes neurons, microtubules are formed when tubulins are stimulated to assemble by tau protein. [62] With a stoichiometry of 9–10 units of phosphatase per mole of protein, the phosphor protein tau is abnormally hyperphosphorylated in the brains of people with Alzheimer's disease (AD). Tau typically contains two to three phosphates per molecule. In AD hyperphosphorylated tau, over 30 sites of phosphorylation have been identified, including ones that they were not phosphorylated in typical tau.[63] In order to better understand the role of aberrant tau phosphorylation in spindle disruption in the AD brain, it looked at the capacity of both typical cytosol tau and AD P-tau to bind to microtubule and promote microtubule assembly, as well as the impact of alkaline phosphatase therapy on microtubule assembly.[64] The human central nervous system has six main isoforms of tau, a highly soluble protein that is naturally disordered. Tau isoforms with 0, 1, or 2 inserts in the N-terminal projection domain (N1 and N2 isoforms) and 3 or 4 pseudo-repeats (3R and 4R isoforms) in the Tau repeat domain (RD), which covers the majority of the MT-assembly domain1 and is in charge of Tau aggregation, are produced by the different isoforms' contents of three alternatively spliced exons. (Figure 8)[65] Phosphorylation-dependent antibodies recognize PHF tau but not recombinant or normal adult tau. This makes it possible to identify areas that are incorrectly phosphorylated. According to the longest human cerebral tau isoform, antibodies AT8 needs phosphorylated of Ser-199 and/or Ser-202, whereas antibodies T3P and PHF1 detect phosphorylated Ser-396. [66]
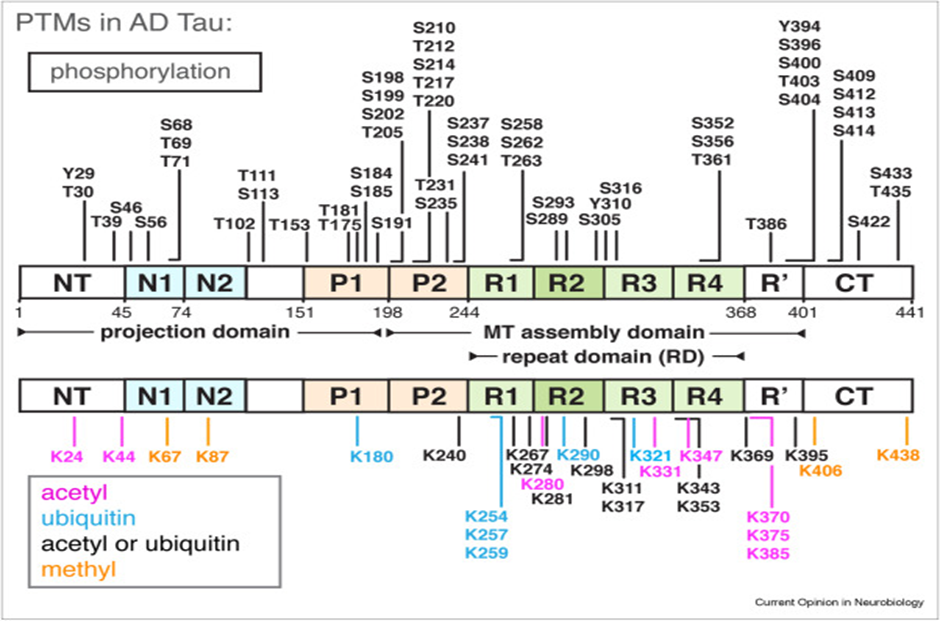
Figure 8 the RD and its adjacent flanking domains [65]
The neuronal protein tau is primarily seen in CNS dendrites. The Src kinase Fyn is transported to the dendrite compartment by Tau. After that, Fyn binds with PSD95 and phosphorylates NMDA. Research on APP transgenic (APP t g) APP23 mice demonstrates that the PSD95 complex and A? result in higher excite toxicity, mortality, and memory deficits. Tau transgenic mice with reduced rapid axonal transport and microtubule density exhibit amyotrophy, axonopathy, and neurological disorders. [66] Phosphorylation-dependent antibodies recognize PHF tau but not recombinant or normal adult tau. This makes it possible to identify areas that are incorrectly phosphorylated. According to the longest tau isoform found in the human brain, antigen AT8 involves phosphorylated of Ser-199 and/or Ser-202, whereas antibodies T3P and PHF1 detect phosphorylated Ser-396. [67]
Tau aggregation
In the human brain, tau aggregation disorder spreads in a very identifiable and predictable way. The entorhinal cortex's layer II is where it begins. The perforant route then allows the illness to spread to the hippocampus. The entorhinal cortexes layer IV and other limbic regions receive projections from the hippocampus. The disease then moves into the isocortex, passing initially through the parietal and temporal lobes before reaching its frontal and occipital neocortex. This progression and spread pattern serves as the foundation for the 6-stage Braak staging system for neurofibrillary degradation in Alzheimer's disease. There are three stages in Braak's stages for β-amyloid deposition: increasing amyloid (stages A–C) and no deposits. According to a study of 2661 autopsy cases involving people aged 25 to 95, tau aggregation happens about 30 years before beta-amyloid plaques, which confirms earlier findings.[68]
Phosphorylated Tau
AD often leads to the loss of neurons and their connections in the entorhinal cortex, hippocampus, and cortex responsible for language, behavior, and thinking. Phosphorylated tau and amyloid beta accumulation in the brain impair synaptic function and cause neuronal cell death. Modifiable and non-modifiable risk factors that lead to AD are listed in Figure 7. [69]
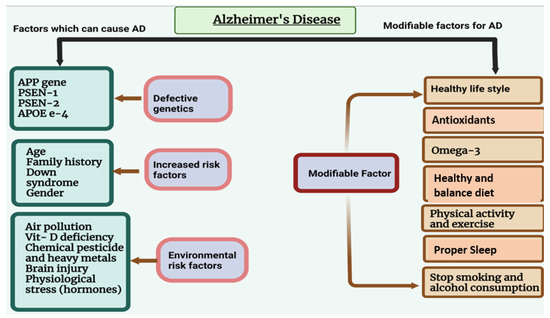
Figure 9 Alzheimer's disease risk factors that can be changed and those that cannot. AD may be caused by non-modifiable factors such as age, sex, and genetic defects. As preventive measures, modifiable risk factors—such as obesity, smoking, alcohol use, heart disease, and physical inactivity—can be very important
This protein can be found in the cytosol, cell membrane, and axons. Tau protein appears as a succession of polypeptides with varying lengths on electrophoresis gels, indicating different RNA splicing and/or phosphorylation levels. The tau gene has at least 16 exons and is located on chromosome 1.[70] 79 of the 441 residues in the largest tau version in the central nervous system, which has drawn the most attention, can be changed by the serine/threonine phosphorylation of tau. Both proline-directed kinases, such GSK3, cdk5, p38, or JNK, and non-proline-directed kinases, like PKA, PKC, CaMKII, MARK, or CKII, phosphorylate at least thirty serine/threonine residues in AD. Moreover, tau binding to microtubules seems to be modulated by this phosphorylation. Of the kinases listed above, GSK3 plays a crucial role in regulating tau phosphorylation under pathological conditions. In fact, sure animal models have demonstrated a connection between aberrant tau aggregation formation and tau phosphorylation by GSK3. There is more proof that tau that has been pseudohyperphosphorylated damages cells and encourages apoptosis. In fact, phosphorylated tau may build up in neurons and negatively impact the cell since it seems to be resistant to being broken down by various proteases. [71] P-tau and t-tau levels are higher in AD patients than in untreated controls, and the biomarker amounts increase as the illness worsens. One excellent method for differentiating AD with healthy controls is to use CSF t-tau. P-tau and t-tau levels are higher in AD patients than in healthy people. Controls, in which the biomarker concentrations increase in direct proportion to the disease's severity. The CSF t-tau is excellent. To differentiate AD from healthy controls. [72] Another technical challenge is the low concentration of tau in CSF, which varies from roughly 300 ng/L in healthy individuals to 900 ng/L in AD patients. Since this quantity is distributed throughout many modified forms and six splice variations, the amount of each molecular species that is available for examination is close to the detection limit of most procedures. [73]
Assays
In the solid-phase enzyme immunoassay called the INNOTEST PHOSPHO-TAU assay, P-tau protein is captured by the monoclonal antibody (mAb) HT7 (Innogenetics, Gent, Belgium). 75 mL of undiluted CSF is mixed with 25 mL of the biotinylated anti-P-tau mAb AT270 (Innogenetics), and the mixture is then incubated overnight at 2–8 °C. Any antigen-antibody complex that has developed is then found using peroxidase-labelled streptavidin (SV-PO). Two wash phases are specified in the test instructions: one after SVPO and the other after the antigen and biotinylated identification antibody have been incubated. An automatic and human wash method did not significantly vary on a Comparable instrument (data not shown). The assay includes a sample addition measuring device to confirm the presence of CSF or SV-PO. The phosphorylated peptide used for the calibration contained the epitopes of HT7 (epitope P159PGQK, numbered according to the longest tau isoform) and AT270 (epitope P176PAPKTP; molecular weight, 3455 Da; peptide sequence, acetyl-P154RGAAPPGQKGQANATRIPAKTPPAPKTPPSSGE; obtained from Neosystems, Strasbourg, France) (7). The calibrator peptide should be regarded as a benchmark. It differs from the normal P-tau protein in terms of size and structure. The format is used to quantify all tau isoforms that are phosphorylated at threonine 181. [74]
Treatments of Alzheimer’s Disease
However, the absence of successful disease-modifying drugs that emerged from this research illustrates how challenging it is to develop a therapeutic agent that could alter the course of a condition as complicated as AD. Over the past ten years, a significant amount of research on AD has been devoted to disease-modifying therapies, which aim to alter the progression of the illness rather than only treat its symptoms. [75]
Cholinesterase inhibitors
In December 2000, the FDA authorized rivastigmine, donepezil, and tacrine as symptomatic therapies for mild to moderate Alzheimer's disease.Tacrine is regarded as the 'first generation' ChEI. Newer medications, like donepezil, offer pharmacological or pharmacokinetic advantages that make them easier to administer and have fewer adverse effects than older ones. Tacrine has been connected with liver damage.[76] The choline acetyl transference enzyme, which changes choline into the neurotransmitter acetylcholine, is significantly less active in the cerebral cortex and hippocampus of Alzheimer's patients.[77] Direct cholinergic agonists and precursor loading have not worked well or been poorly tolerated. Cholinesterase inhibitors (ChEI), on the other hand, have been shown to be effective and to have a manageable adverse effect profile. By blocking acetylcholinesterase, which degrades acetylcholine, these drugs increase the amount of acetylcholine that may interfere with postsynaptic receptors. AD therapy was made possible by the first tacrine that accumulate trial, which was published fifteen years ago.[77] Cholinesterase inhibitors have been shown through experimental research to have a variety of additional pharmacological and biochemical actions in addition to their effects on synaptic acetylcholine, which may contribute to their therapeutic efficacy in AD patients. These activities include binding to nicotinic receptors' allosteric activator site, enhancing the release of noncholinergic neurotransmitters, preventing β-amyloid toxicity, boosting the release of soluble amyloid precursor proteins, and adjusting the effects of estrogens. Cholinesterase inhibitors' nonhydrolytic effects, particularly their potential association with β-amyloid, are clinically significant. Still being assessed in AD patients.[78]
Tacrine
Tacrine, a small molecule medication, is the first licensed cholinesterase inhibitor (ChEI) for treating Alzheimer's disease. Other approved ChEIs include donepezil, rivastigmine, and galantamine. Tacrine has been linked to certain adverse effects. In recent years, researchers have attempted to improve the efficacy of tacrine by synthesizing hybrid molecules referred to as ChEIs. As per figure 9[79]
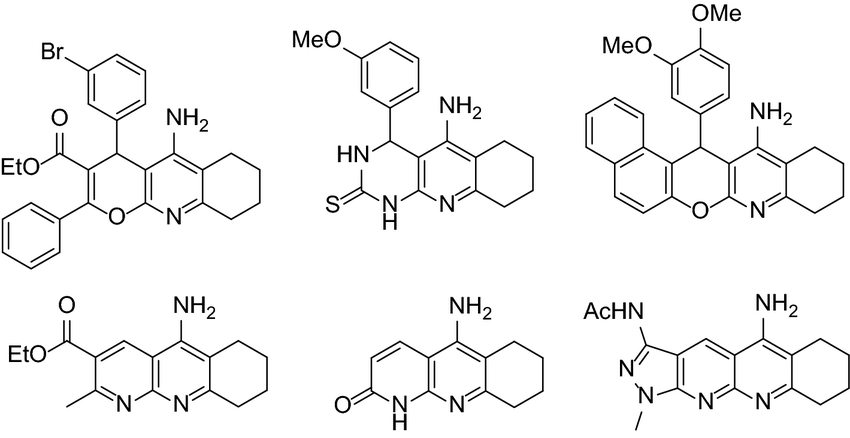
Figure 9. Tacrine-based hybrids as ChEIs
Memantine
Due to the significant symptomatic effect that memantine has demonstrated in many large, controlled clinical studies, it is recommended for the treatment of people with moderate to severe AD.Memantine is an uncompetitive inhibitor of the N-methyl-D-aspartate (NMDA) transporter with a moderate affinity. Memantine was created in 1968, and Eli Lilly received a patent for it. It was later developed by Merz in collaboration with Neurobiological Methods, Inc. A license for building it in the US and the Company for other international markets was then given to Forest Laboratories. Memox® (Unipharm), Namenda® (Forest Laboratories), Ebixa® and Abixa® (Lundbeck), and Axura® and Akatinol® (Merz) are the proprietary brands for memantine.[80,81]Memantine has drawn a lot of interest despite only recently being authorized for the treatment of moderately to severe Alzheimer's disease, also called AD, in both the US and Europe. Nonetheless, memantine may have a far broader range of therapeutic uses and is helpful in treatment other dementias and diseases of the brain like Parkinson's and Huntington's disorders. [82]
Memantine's mechanism of action in AD treatment
Similarly, memantine may temporarily moderate the cognitive and behaviour decline and also produce some enhancement over the first three months of treatment. The five neuropathological features of AD include intraneuronal neurological tangles (NTFs), excitotoxicity of glutamate, exogenous amyloid-β accumulation (Aβ plague), acetylcholine deficiency, and neuroinflammation. Memantine treatment for AD is associated with excitation toxicity of glutamate and cholinergic deficiency. In AD patients, there is an increase in acetylcholinesterase activity, acetylcholine breakdown, and acetylcholine levels in the brain. By encouraging central brain neurotransmission activity, increasing brain activity and excitement, and favourably affecting memory encoding, consolidation storage, and retrieval processes, acetylcholine improves memory. [83, 84] Additionally, memantine protects neurons against glutamine-induced excitotoxicity. Additionally, memantine cannot slow the progression of the disease, stop neuronal death, or stop dementia from becoming more serious. Memantine is commonly recommended in the early stages of AD after receiving regulatory permission .Enhancing cholinergic signalling is intended to alleviate cognitive deficiencies and memory impairment in neurodegenerative diseases, such as AD. Acetylcholine neurotransmission is essential for learning and memory. [85]. Memantine is often used at a dose of 20 mg/d to treat AD. Memantine at doses of 5–30 mg/d was administered, and two to three hours later, human cerebrospinal fluid levels of 0.05–0.31 μM were detected.[82] Memantine has been demonstrated to improve global cognition, functional communication, and a few behavioral symptoms, such as agitation and aggression. It's interesting to note that donepezil and memantine have different behavioral effects; donepezil affects sadness, anxiety, and apathy, while memantine mostly impacts agitation, aggression, and delusions. A recent clinical research suggests that using donepezil and memantine together to treat moderate-to-severe AD may be secure and well-tolerated. But when compared to donepezil alone, donepezil plus memantine does not considerably enhance cognitive function. Combining treatment might therefore be more effective in improving neuropsychiatric behaviors than cognitive performance due to their complementary activities.[86]
Cerebrolysin
In several European and Asian countries, cerebrolysin, another non-cholinergic treatment, is approved for AD and other dementias. This drug is made from refined neurological proteins that has been through enzymes broken down to create single amino acids and active small peptides that can cross the blood-brain barrier.It has neurotrophic effects that are comparable to those of natural nerve growth factors (NGF), specifically supporting and protecting neuronal function. [87] The exact mechanisms by which cerebrolysin works as a treatment are still unknown, though. Studies have shown associations with amyloid peptides, inflammatory processes, regulating receptors for neurotransmitters (adenosine A1 and GABAB), and neurotrophic proteins such as glycogen synthase kinase-3 (GSK-3) and cyclin-dependent kinase-5 (CDK-5).For 10–20 days, 5–30 mL of cerebrolysin per day is the recommended intravenous dosage.[87]
Reducing Aβ Production
The two proteases that change APP into Aβ are β-secretase and γ-secretase. α-secretase, a third protease, competes with β-secretase for the APP substrate, preventing the formation of Aβ. In order to lower Aβ, three methods have been suggested: β-secretase inhibition, γ-secretase inhibition, and α-secretase stimulation. [88]
B-Secretase suppression
Two proteolytic breaking down events are needed to create Aβ from its precursor: one at the the C-terminus by γ-secretase and one at the end of the tail by ?-secretase. There are many advantages of using secretase as a therapeutic target for ads. The absence of an in-secretase gene deletion indicates that it first starts the synthesis of A and lacks compensatory activity. [89, 90] A vital enzyme that is identified early in the sequence of biological processes that result in the development of disease is β-secretase, also known as the membrane correlated aspartic the protease 2 (memapsin 2), beta-site precursor protein for amyloid breaking down enzyme 1 (BACE1), or aspartyl protease enzyme 2 (Asp2).Small molecule inhibitors have made BACE1 an intriguing therapeutic target that may change how Alzheimer's disease progresses. [91] Furthermore, dopaminergic failure and memory and learning problems shown in a transgenic mouse model for AD may be prevented by BACE1 deficiency [88].
Γ-Secretase suppression
Presenilins (PS1 and PS2), these have been found to be predisposing mutations in individuals with familial dementia (AD), are thought to contain the g-secretase active site. The activity of g-secretase is one of the few biological targets for drugs that will lower the quantity of amyloid plaque, whose accumulation is thought to cause AD.The proteolytic activity known as secretase is in charge of cleaving a number of essential membrane proteins. [92, 93] Though the mechanism requires further investigation, γ-Secretase may be classified as an aspartyl protease. PS are produced as precursor proteins that must be integrated into a bigger complex for stability. The pool that is not incorporated into these complexes is destroyed quickly by the proteasome. PS stabilization is accompanied by a proteolytic "maturation" cleavage done by an unknown "presenilinase." This information is crucial for calculating the lowest number of subunits required to form the γ-Secretase complex. [93, 94]
Table 1- Typical signs of AD include agitation and psychosis.
|
Agitation |
Psychosis |
|
Repetitive mannerisms, hoarding, screaming, hitting, wandering, verbal aggression, and general restlessness |
Delusions concerning theft, harm and spousal infidelity; visual hallucinations. |
The fact that a number of novel drugs are being investigated for agitation in AD is heartening; nonetheless, the number for psychotic is far smaller. In addition, we are learning more about the biology that underlies these symptoms. Given these two factors, it is likely that the use of traditional antipsychotics to treat aggression and psychosis in AD will be reexamined. Several other investigations found that tau hyperphosphorylation was associated with psychopathology in women but not in males. This is in line with previous research showing women have a greater chance than men to have psychosis, as well as data from cerebrospinal fluid examination and neuroimaging. The chronology of events may be clarified by longitudinal PET examination of tau, as there is still ambiguity over whether the pathology of tau causes madness or is associated with another mechanism. Higher levels of white matter binding were linked to more severe late-onset psychosis in a research using the tau PET tracer 11C-PBB3 in patients with trauma to the head, but this study has not yet been done in AD psychosis. This conclusion would suggest that additional in vivo study on the relationships among tau and AD psychosis is required, especially when combined with results from post-mortem investigations. [96]
Anti-Tau Therapy
TRx0237 (LMTX) is one tau aggregation inhibitor. It decreases the quantity of aggregated tau proteins to lessen tau-related neuronal damage. A TRx0237 experiment was carried out from October 2012 to May 2016 to investigate the drug's efficacy in treating mild AD. The main indicators of outcome for the clinical trial were alterations to the performance of two scales: the ADAS-cog 11 and the ADCS-ADL 23.TRx0237 did not work as an adjuvant treatment for AD, according to the trial's results. TRx0237's continuous phase 3 trial got underway in January 2018. The goal of this study is to determine how well TRx0237 works in people with early-stage AD at different dosages. The primary outcome measure is the shift in the standard uptake value ratio according to temporal lobe 18Ffluorodeoxyglucose PET. This experiment is scheduled to run through December 2020. The active form of vaccine AADvac1 stimulates the immune system by targeting numerous critical epitopes in pathogenic forms of tau, which stops tau from aggregating and lessens the formation of neurofibrillary tangles. [98]The ways that an immunotherapy might be able to help are highlighted by our present understanding of how tau disease spreads. Initially, pathogenic tau is produced intracellularly in pre-aggregate forms. Targeting this phase would require antibodies to be capable to enter neurons, which may or may not be possible. The affected neuron begins to emit tau seeds, also known as tauons, when intracellular disease reaches a particular stage. [97, 98, 99]
CONCLUSION
Alzheimer’s disease presents a formidable challenge to modern medicine due to its complex etiology, insidious onset, and lack of curative treatments. Even after decades of study, the exact cause of the illness is still unknown, but tau hyperphosphorylation and amyloid-β buildup are important factors in its pathophysiology. The complex character of AD is further highlighted by the presence of oxidative stress, mitochondrial dysfunction, metal ion imbalance, neuroinflammation, and neurotransmitter dysregulation. Early diagnosis and staging have been improved by the discovery of trustworthy biomarkers including CSF Aβ42, T-tau, and P-tau as well as imaging techniques like PET scans. This has created a window for possible intervention before irreparable damage is done. The significance of individualized risk assessment and prevention strategies is further highlighted by risk factors like genetic mutations (APP, PSEN1, PSEN2), APOE ε4 genotype, comorbidities like diabetes, hypertension, and cardiovascular diseases, and modifiable lifestyle factors like diet, physical activity, and smoking. Cholinesterase inhibitors and memantine, two currently authorized therapies, relieve symptoms but are unable to stop or reverse the course of the disease. Clinical studies investigating newer treatment options, such as lowering tau aggregation, modifying glutamate excitotoxicity, managing neuroinflammation, and targeting Aβ generation using β- or γ-secretase inhibitors, have shown mixed results. In summary, the most promising strategy is a comprehensive one that incorporates targeted multimodal medicines, lifestyle changes, early detection, and precision diagnostics. Finding disease-modifying therapies and, eventually, a cure for Alzheimer's disease will need sustained cooperative research efforts.
ACKNOWLEDGEMENT
We would like to express our heartfelt gratitude to all those who supported and guided us throughout the completion of this article. First and foremost, we sincerely thank Dr. Shivajirao Kadam College of Pharmacy for providing us with the necessary facilities and a conducive academic environment. We are deeply grateful to all the faculty members of the institution for their constant encouragement, valuable suggestions, and continuous support during the preparation of this work. We would also like to extend our special thanks to our mentors and guides for their insightful guidance, motivation, and constructive feedback, which helped us improve the quality of our work. This article is a collaborative effort by the following authors: Manali Bavadekar, Avadhut Khot, Vaishnavi Kolekar, Sanika Kokate, Aishwarya Patil, Vaishnavi Bagal, Aditya Jagtap, Alfaj Jamadar, Aniket Gavali. Their dedication, teamwork, and commitment to research have been instrumental in the successful completion of this article.
REFERENCES









Manali Bavadekar*, Avadhut Khot, Vaishnavi Kolekar, Sanika Kokate, Aishwarya Patil, Vaishnavi Bagal, Aditya Jagtap, Alfaj Jamadar, Aniket Gavali, Neurodegenerative Cascade A Part of Alzheimer's Illness - Multifactorial Dissection in Amyloidopathy, Tauopathy, And Neuroinflammatory Crosstalk, Int. J. of Pharm. Sci., 2025, Vol 3, Issue 8, 1098-1126. https://doi.org/10.5281/zenodo.16795722
 10.5281/zenodo.16795722
10.5281/zenodo.16795722