We use cookies to make sure that our website works properly, as well as some ‘optional’ cookies to personalise content and advertising, provide social media features and analyse how people use our site. Further information can be found in our Cookies policy
Iohexol, a tri-iodinated contrast agent, plays a crucial role in diagnostic imaging, computed tomography (CT) and renal function assessment. Its accurate quantification in pharmaceutical formulations is essential to ensure product quality, efficacy, and patient safety. This review provides a comprehensive overview of various analytical techniques employed for the qualification and quantification of iohexol in pharmaceutical preparations. Methods such as high-performance liquid chromatography (HPLC), ultra-performance liquid chromatography (UPLC), and spectrophotometry. Additionally, recent advancements in analytical methodologies, including hyphenated techniques like LC-MS) and tandem mass spectrometry (LC-MS/MS) is highlighted for their enhanced capabilities. The review also explores the challenges associated with iohexol analysis, including sample preparation, matrix interferences, and method validation parameters such as accuracy, precision, linearity, limit of detection (LOD), and limit of quantification (LOQ). A comparative analysis of the existing techniques aims to guide researchers and pharmaceutical analysts in selecting the most suitable method for precise iohexol quantification.
Iodinated X-ray contrast media (ICM) are tri-iodinated substances that improve the contrast between organs and surrounding tissues, allowing for the visualization of organ features that would otherwise be impossible to study. ICM is one family of medicines. They are used at far larger dosages than any other intravascular medication, and they are consumed globally at a rate of about 3.5 106 kg1.
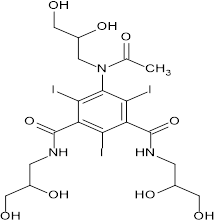
Iohexol is an iodinated radiographic discrepancy medium for use in individual radiology. Its concurrence from the tube following a single injection can be used to estimate the glomerular filtration rate (GFR) in humans and domestic animals because it is not protein-bound and is eliminated by the body through glomerular filtration2. Iohexol can be quantified by different methods, including X-ray fluorescence (XRF), Liquid chromatography-tandem mass spectrometry (LC-MS/MS), Mass Spectroscopy, Ultra-performance liquid chromatography (UPLC), and High-performance liquid chromatography (HPLC), Capillary Electrophoresis (CE), High-Performance Liquid Chromatography with Ultraviolet Detection (HPLC-UV)3. One of the labels that is frequently used to determine mGFR is iohexol. It has been said to be appropriate for both adults and children to have an accurate GFR measurement. This X-ray discrepancy medium is low osmolar, non-ionic, and chemically stable. Its monoisotopic molecular mass is 820.9 Da. Iohexol administered intravenously is simply removed from the tube by glomerular filtration. It has been demonstrated that the order does not exhibit tubular stashing, metabolism, or reabsorption of iohexol. Similarly, iohexol has a protein list that is less than 2. Originally, iohexol was employed in urographic and angiographic operations4.
Iohexol
A non-ionic, low molecular weight iodinated compound, iohexol (IOH) is 5-[acetyl(2,3-dihydroxypropyl) amino]-1-N,3-N-bis(2,3-dihydroxypropyl)-2,4,6-triiodobenzene-1,3 dicarboxamide. With the ratio of both of the isomers remaining constant, it displays endo exo stereoisomerism concerning the substituted aromatic ring. The chemical is frequently employed as a contrast agent for procedures like computed tomography, catheter-based angiography, and others. In terms of its pharmacokinetic characteristics, IOH has a modest protein binding of 1.5% and is widely dispersed. IOH has a very limited extra-renal clearance (less than 5%), and it is neither produced nor absorbable at the tubular level. Since IOH is not nephrotoxic and has a low osmolality, it is usually regarded as safe. There have been no reports of serious side effects or anaphylactic reactions anywhere in the world5.
Mechanism of Action: Natural iodine compounds piece x-rays as they pass through the body, subsequently permitting body structures containing iodine to be depicted in differentiate to those structures that do not contain iodine. The degree of opacity created by these compounds is specifically relative to the add up to sum (concentration and volume) of the iodinated differentiate specialist in the way of the x-rays. After intrathecal organization into the subarachnoid space, dissemination of iohexol in the CSF permits the visualization of the subarachnoid spaces of the head and spinal canal. After intravascular organization, iohexol makes misty those vessels in its way of stream, permitting visualization of the inner structures until critical hemodilution happens.
Analytical Review of Iohexol:
Liquid chromatographic methods for assay of IOHEXOL were reported in United States Pharmacopeia using Acetonitrile and Water as mobile phase. Iohexol detected at UV 254 nm. The Flow rate is 1.0mL/min. and the retention time in range of 0.84 to 1.0 min. Several analytical methods for determination of Iohexol were published including the following.
Hyphenated Techniques:
Liquid Chromatography - Mass Spectrometric (LC-MS)
Anna Schulz et.al worked on LC- ESI- MS analyses performed using an Agilent 1200 series liquid chromatographic system, supplied an electrospray ionisation source (ESI) for the HTC mass spectrometer. This column (150 mm × 0.5 mm, 5µm) is a Zorbax SB C18 column. The temperature of the column was kept at 50 °C. 0.1 Formic acid in water (v/v) (A), on and 0.1 formic acid in acetonitrile (v/v) (B) were the mobile phases. The rate of flow was 50 µl/min. It ran for sixteen mins. 4,000 V served as the ion spray voltage. The drying gas flow speed reached 8 l/min, and the dry temperature was 300 °C. 20 psi was the nebuliser gas setting. Iohexol was utilised to spike the lower quality control (LQC) at 200 pg, the middle quality control (MQC) at 2 ng, and the highest-quality control (HQC) at 20 ng. A linearity range of 50 pg–40 ng was revealed by the validation. The range of 2.7–12.1 was determined for the intraday and interday precision. The accuracy ranged from 91.2 to 98.7 both within and between days. 100% was the mean recovery rate4.
Julianne L. Holleranet.al investigated the use of high-performance liquid chromatography mass spectrometry to quantify iohexol in 50 L of human plasma. Shodex Asahipak NH2P-50 2D column, 5 m, 2 × 150 mm. A Micromass Quatromicro triple-stage benchtop mass spectrometer with electrospray and positive-mode ionisation was used for mass spectrometric detection. 0.1% formic acid in acetonitrile and 0.1% formic acid in water made up mobile phase solvents A and B, respectively. Run time: 10 minutes. The iohexol assay demonstrated linearity from 1 to 500 μg/mL and demonstrated accuracy (101.3–102.1%) and precision (<3.4%CV). Retention time of iohexol was 3.6 min. The range of QC based accuracies was 101.3–102.1%. Recovery from plasma was 53.1–64.2 % and matrix effect was trivial (−3.4 to −1.3 %). Plasma freeze thaw stability (97.4–99.4 %), stability for 5 months at −80 ?C (95.5–103.3 %), and stability for 4 h at room temperature (100.6–103.3 %) were all acceptable. This validated assay using a deuterated internal standard will be an important tool in measuring iohexol clearance and determining glomerular filtration rate (GFR) in patients11.
Vicente Faye B. et al.An LC-MS/MS analysis was performed using the analytical column Luna C8 (3 μm, 50 × 3.0 mm) and a Waters 2795 Alliance HT HPLC system coupled to a Waters Corporation Micromass Quattro Micro tandem quadrupole mass spectrometer functioning in the electrospray positive ion mode with multiple reaction monitoring (MRM). Two milligrammes of ammonium acetate and 0.1% formic acid in water and methanol, respectively, were introduced to mobile phases A and B while maintaining a constant flow rate of 0.500 mL/min. The retention durations for iohexol and ioversol were 2.36 and 2.14 minutes, respectively. The solvent flow was redirected from its point of origin to waste at 0–1 and 5–7.5 minutes. Iohexol has a linear test range of 7.7 to 2000 μg/mL. Iohexol's detection limit was 3.0 μg/mL. The LLOQ was 7.7 μg/mL, with a linear recovery rate of 97%. The intra-day precision for serum-based QC samples is from 1.0 to 3.8%, while the inter-day precision is 1.5 to 3.9%. The average accuracy values for the various concentrations varied from 2.1 to 4.4% and 2.2 to 5.0%. A Deming slope of 1.030 and an intercept of −2.408 were found using regression analysis. After triple freeze-thaw cycles, serum samples kept for 24 hoursat 4 °C, 1 month at -20 °C, and 3–4 months at -20 °C show a 3% change in mean values. This technique accurately measures iohexol in 50 μL of serum. MRM-monitored ion transitions included iohexol (m/z 821.9 → 803.7) and ioversol (m/z 807.9 → 588.7).12
Soo-Youn Lee et.al developed a simple, fast, and accurate HPLC-MS/MS method to determine the concentration of iohexol in serum. Waters 2795 Alliance HPLC is a part of the Quatromicro API tandem mass spectrometer. An Eclipse XDB-C8 column (100 mm × 2.1 mm i.d., 3.5 m) was utilised. The mobile phases were 2-mM ammonium acetate and 0.1% formic acid in water (A) and 2-mM ammonium acetate and 0.1% formic acid in methanol (B). Serum samples were combined with I.S. (bromperidol) and centrifuged for three minutes following basic protein precipitation using ZnSO4. Three degrees of extraction recovery were obtained, ranging from 94.6 to 107.4%. To do quantitative analysis, the multiple reaction-monitoring mode (m/z 822.0 → 804.0 for iohexol, 420.1 → 122.7 for I.S.) was employed, and each sample ran for three minutes. R2 > 0.997 indicated that the assay was linear between 0.5 and 1500 μg/mL. There were 2.4–6.2% and 5.5–6.5% intra- and inter-assay coefficients of variance, respectively.13
Chromatographic Methods.
High Performance Liquid Chromatography (HPLC).
Soufiane El Assri et al. developed an HPLC-UV method for measuring iohexol concentrations in blood and urine, improving analytical tools for assessing kidney function.The HPLC was performed utilizing a LC-2030C Prominence-I Shimadzu instrument. The three sorts of explanatory columns utilized: VDS Optilab column (150 × 4.6 mm; 5 μm molecule estimate C18) from Chromatographie Technik Gmbh (Berlin, Germany), Lichrospher explanatory column (250 × 4 mm; 5 μm molecule measure) and Lichrospher expository column (100 × 10 mm; 5 μm molecule measure). Discovery was accomplished with the RF-20A locator at an absorbance wavelength of 254 nm.The flexible stage consists of water and 5% acetonitrile (95:5 v/v), a C18 Lichrospher expository column (250 × 4 mm, 5 μm molecule estimate), and a 40°C temperature and a flow rate of 1 ml/min. The serum steadiness proportion of iohexol is between 97.84 and 99.04% with a non-significant variety. In the urine, the proportion changes between 97.63 and 99.31% with a non-significant variety. For urine a recuperation rate <95% for low concentration14.
Hu Nana created a High-Performance Liquid Chromatography system applying a diode array detector, an HPLC column (RP 18e 100 x 4.6 mm), and a guard column (RP-18e 5 x 4.6 mm). Iohexol was eluted with acetonitrile/water (4:96%) at a flow rate of 1.5 ml/min and kept at 30°C for a total run time of 4 min. The UV absorbance was monitored at 254 nm. Iohexol had a retention time of 2.5 minutes. The eluting time was 4 min. An iohexol standard solution at 291 μg/ml separated by HPLC. The calibration curve of the HPLC method was constructed in the range of 23.29 ? 291.15 μg/ml. The HPLC method's intra-day iohexol quantification accuracy ranged from 90.6 to 108.8%. The inter-day accuracy was 96.93–98.25%. The precision of intra-day was 0.1–5.1%. The precision of inter-day was 2.27–4.86 % HPLC method, the coefficient of variation never surpassed 5%, and the accuracy was within 10%3.
Giannakis Stefanos et al.Using an HP Agilent 1100 Series HPLC analyser with a G1315A diode array detector, the concentration of iohexol was determined. The mobile phase, which included a combination of 95% ultrapure (Mili-Q) water, 0.1% formic acid (phase A), and 5% methanol (phase B), was maintained in an isocratic mode throughout the whole analysis. There is a 1 mL/min flow. A temperature column of 40 C was used. A 50 mL injection volume was used. Retention time was 10.15 min. The wavelength of the UV detector is 254 nm15.
Harvey A. Schwertner et al. developed a chromatography technique to identify iohexol standards, plasma iohexol calibrators, and plasma test samples on a Waters Chromatographic System that included a Separations Module, Photodiode Array Detector (PDA), and a Millennium 32 Chromatographic Manager. A C-18 reversed-phase column was used for the chromatographic analysis. Trifluoroacetic acid (pH 2.2)–methanol (80:20, v/v) at 0.1% is the mobile phase. Chromatographic analysis was performed on a C-18 reversed-phase column. 0.1% trifluoroacetic acid (pH 2.2)–methanol (80:20, v/v) is the mobile phase. The HPLC operating parameters were as follows: injection volume was 5.0 Ml. Column flow rate was 0.3 mL/min. Chromatographic run time was 16.0 min. PDA spectra recording at 245 nm. UV absorbance of iohexol (0.01 mg/mL) in 0.1% TFA (pH 2.2)–methanol (80:20, v/v) and in water.UV scans of iohexol were carried out in both the HPLC mobile phase (0.1% TFA, pH 2.2)-methanol (80:20, v/v) and water. The limit of detection is 6.0 μg/ml. The lower limit of quantification was 10.0 μg/ml. Accuracies between 89.0-108.4%16.
Fatemeh Akhlaghi et.al developed and validated an HPLC-UV analytical approach to estimate the exact amount of iohexol within human plasma. Protein precipitation and iohexol recovery from 100 μl of plasma were performed using 800 μl of 5% v/v perchloric acid and iohexol-related molecule B as an internal reference. Vortex mixing and centrifugation were then performed. The supernatant (90 μl) was then injected onto μBondapak C18 column (150mm×3.9mm, 10 μm) maintained at 30?C. The mobile phase acetonitrile and water. A total run time of 12min. The wavelength for maximum UV absorption of iohexol (λmax) was found to be 254 nm. The assay was linear from 10-750 μg/mL of iohexol, having the mean correlation coefficient (r2) of 0.999 (n=8). The accuracy of the estimated iohexol concentration was more than 90% at three QC concentrations. The precision expressed as inter-day coefficient of variation (CV %) ranged from 1.6% to 3.2% and the intra-day CV% ranged from 0.5% to 3.7%. The limit of detection and limit of quantification for iohexol were 6 and 10 μg/mL, respectively. A simple, quick, and dependable HPLC-UV analytical technique for estimating iohexol utilising a readily accessible internal standard17.
Thotsaphorn Chaloemsuwi wattanakan et.aldeveloped Micro-liquid chromatographic system (μ-LC) consisted of a high-pressure pump Model LC–10AD equipped with six-port valve micro injector with 250nL sample loop Model C2N-4346. UV-Visible detector Model Spectro flow 757. Fused-silica capillaries with 100 μm id, 365 μm OD were obtained from Agilent. An HPLC instrument Model 1100 (Agilent, USA) with 5 m Hypersil RPEP C8 Column (150 mm × 4.6 mm, Thermo Scientific, UK) was used for method comparison. A mobile phase of 40:60 v/v 50 mM phosphate buffer pH 5: methanol. UV-detection at 254 nm, flow rate 2 L/min. Calibration curves in the concentration range of 2-500mg/L. Theoretical plate number of 48226. The limit of detection was 0.44 mg/L whereas the limit of quantification was 1.47 mg/L18.
Etienne Cavalier et.al worked on HPLC equipment consisted of a Hewlett–Packard (HP) 1100 Model. The Lichrospher analytical column (250×4 mm I.D.; particle size: 5 μM) was packed with C18 material. The mobile phase consisted of a mixture of distilled water and acetonitrile (95:5: v/v) adjusted to pH 3.0 with orthophosphoric acid. The separation was performed isocratically with a flow rate of 1.0ml/min. Typical HPLC operating pressure was approximately 70bars at a thermostatised column oven temperature of 40 °C. The HP 1100 autosampler was utilized for to inject an amount containing 20 μl of serum as well as urine. Detection was achieved with the HP 1100 DAD detector at an absorbance wavelength of 254 nm. The flow rate 1 ml/min. The limit of detection were 3.08 μg/ml for serum and 26.06 μg/ml for urine. The quantification limits for serum were 10.76 to 1295 μg/ml, while for urine, they were 86.0 to 4144 μg/ml19.
Domenic A. Sica et.al reported a simple HPLC method was developed for the simultaneous determination of iohexol, iothalamate, p-aminohippuric acid (PAH) and n-acetyl-p-aminohippuric acid (n-acetyl-PAH) in human plasma and urine. There was a Hewlett-Packard (HP) Model 1090 HPLC among the HPLC apparatus. A Supelco Discovery® C18 was used as the analytical column. The mobile phase included a methanol gradient and aqueous trifluoroacetic acid (0.1% TFA in deionised water (pH 2.2). A flow-rate is 1.0 ml/min. At a column oven temperature of 40°C, the average HPLC operating pressure was around 150 bar. An injection volume of 5 μl of the prepared urine sample and 10 μl of the prepared plasma sample was accomplished using the HP Model 1090 auto sampler. UV detector with an absorbance wavelength of 254 nm. Iohexol and iothalamate showed linearity from 10 to 50 μg/ml, PAH from 5 to 40 μg/ml, and n-acetyl-PAH from 2.5 to 40 μg/ml, according to the plasma and urine assay. Accuracy for Iohexol 15.0 to 45.0 μg/ml. The accuracy of all components <8% and the precision within 14%.The absolute recovery for the plasma method was determined to be 100% for all four components. The HPLC plasma and urine results obtained for PAH were used to calculate the subject kidney effective renal plasma flow (ERPF) and the iohexol results were used to calculate the subject kidney glomerular filtration rate (GFR)20.
In this work, Marko Jovanovi? et al. create a hydrophilic interaction liquid chromatographic technique for the Quality by Design (QbD) approach to analyse iohexol, its endo-isomer, and three contaminants. Using the Design of Experiments technique, a link is established between important quality features and critical process parameters, such as the concentration of ammonium acetate in the water phase, the pH of the water phase, and the acetonitrile content in the mobile phase. Plackett-Burman design is used to assess experimental robustness. The analytical column ZIC HILIC (100 mm x 4.6 mm, 5 μm particle size). The mobile phase was composed of acetonitrile and water phase (72 mM ammonium acetate, pH 6.5 with glacial acetic acid) (86.7:13.3) v/v. The column temperature was 25oC. The flow rate being 1 mL/min. The detection wavelength is 254 nm. The linearity range is 0.25–0.75 μg/mL.21.
De Baere, S. et al. reported a protein precipitation stage was used to prepare the sample, which involved mixing 100 L of serum or plasma with 15 L of trifluoroacetic acid. The polymeric PLRP-S column, which measures 250 mm by 4.6 mm, is kept at 30 °C. The gradient elution of water (A) and methanol (B) served as the mobile phase. At a flow rate of 1.0 mL/min, the run lasted 21 minutes. The ultraviolet detector's wavelength was adjusted at 254 nm. For both endoiohexol (0.62–93.0 μg/mL) and exo- (0.44–657 μg/mL), matrix-matched calibration graphs were created. The goodness-of-fit and correlation coefficients ranged from 4.44 to 9.87% and 0.9985 to 0.9999, respectively. The limits of quantification and detection were 0.44 and 0.15 μg/mL for exo-iohexol, and 0.62 and 0.20 μg/mL for endo-iohexol, respectively. The approach is simple, inexpensive, and efficient for detecting exo- and endo-iohexol in feline and canine plasma. It’s usefulness in veterinarian clinical and research applications is shown by its efficient use in pharmacokinetic studies22.
Ultra-Performance Liquid Chromatography (UPLC)
Hu, Nana: Ultra-Performance Liquid Chromatography system with a UPLC column (BEH Peptide 50*2.1 mm, 1.7 µm particles, Waters), a guard column (Acquity BEH C18 2.1*5 mm, Waters), and a diode array detector. Chromatography was performed at 50°C at a flow rate of 1.0 ml/min with a run time of 1.5 min. Solvents A (water, 0.1% formic acid) and B (acetonitrile, 0.1% formic acid) were used to make the mobile phases. UPLC separation of iohexol standard solution at 43.89 μg/ml. The iohexol calibrator at a concentration of 43.89 μg/ml exhibited a retention time of 0.9 minutes. 1.5 minutes was the eluting time. Iohexol's corresponding concentration, which varies between 8.67 and 500 μg/ml. The intra-day accuracy of the UPLC approach ranged from 93.5 to 103.0%. The accuracy between days ranged from 98.7 to 100.6%. Using UPLC, the intra-day accuracy for iohexol analysis ranged from 0 to 7.8%. Inter-day accuracy ranged from 0.8 to 2.5%. At every concentration, the accuracy and coefficient of variation were within 8%3.
Thomas M. Annesley et al.conducted a UPLC-MS/MS analysis utilising a Waters Acquity UPLC system linked to a Waters TQD tandem quad-rupole mass spectrometer operating in the electrospray positive ion mode with multiple reaction monitoring (MRM). A 5-L injection was utilised on a regular basis, while imprecision was tested for 5- and 10-L injection volumes. Chromatographic separation was carried out at 50 °C utilizing a Waters Oasis HLB column (2.1 × 20 mm) with a particle size of 5 μm. The flow rate was 0.4 mL/min. Mobile phase A was 0.1% formic acid in water, and mobile phase B, 0.1% formic acid in acetonitrile. They used a 0.5-min solvent/ divert delay to keep the electrospray probe and sample cone free from buildup of non-retained serum components and collected data beginning at 0.5min. In the concentration range of 2.5 mg/L (the lower limit of quantification) to 1500 mg/L for iohexol, the test showed linearity. The average extraction efficiency achieved was 99%. Recovery rates for the intended target concentrations fell between 99% and 102%. The inter-assay accuracy ranged from 7.9% at 2.5 mg/L to 4.1% at 1000 mg/L. Ion suppression testing indicated no matrix-related interference with the ionization of either compound23.
Table 1: Recent updated research work on Various Analytical method development and validation of IOHEXOL
Sr. No
Author
Method
Description
Year
Ref. No.
1
Hu Nana
HPLC and UPLC
HPLC: - Mobile phase: Acetonitrile / water (4:96%)
Wavelength: 254 nm.
Linearity: 291 μg/ml
UPLC: - Mobile phase: Solvent A (water, 0.1% formic acid) Solvent B (acetonitrile, 0.1% formic acid).
Linearity: 43.89 μg/ml
2023
3
2
Soufiane El Assri
HPLC-UV
Wavelength: 254 nm.
Mobile phase: water and acetonitrile (90:10 v / v)
Linearity: 16 to 1600 μg/ml for serum, 100 to 2150 μg/ml for urine sample
2020
14
3
Julianne L. Holleran
LC-MS/MS
Mobile phase: solvent A 0.1 % formic acid in acetonitrile and solvent B consisted of 0.1 % formic acid in water.
2020
11
4
Stefanos Giannakis
HPLC
Wavelength: 254 nm
Mobile phase: water with 0.1% of formic acid (phase A) and 5%of methanol (phase B).
2017
15
5
Thotsaphorn Chaloemsuwi wattanakan
μ-liquid chromatography
Mobile phase: 40:60 v/v 50mM phosphate buffer pH 5/methanol.
2016
18
6
Marko Jovanovi?
QbD-HILC
Wavelength: 254nm
Mobile phase: acetonitrile – 72 mmol/L ammonium acetate in water adjusted with acetic acid to pH 6.5 (86.7:13.3, v/v)
Linearity: 0.25–0.75 μg/mL
2015
21
7
Faye B. Vicente
LC-MS/MS
Mobile phase: A was 2 mM ammonium acetate and 0.1% formic acid in water and B was 2 mM ammonium acetate and 0.1% formic acid in methanol.
2015
12
8
Anna Schulz
LC-ESI-MS
Mobile phases: 0.1 % formic acid in water (v/v) (eluent A) and 0.1 % formic acid in acetonitrile (v/v) (eluent B). Flow rate: 50 µl/min.
Total run time: 16 min.
The ion spray voltage was set to 4,000 V.
2014
4
9
S. De Baere
HPLC with UV detection
Mobile phase: water (A) and methanol (B) and a gradient elution was performed Acetonitrile.
Wavelength: 254 nm
2012
22
10
Thomas M. Annesley
UPLC-MS
Mobile phase: A 0.1% formic acid in water, and mobile phase B, 0.1% formic acid in acetonitrile
Flow rate: 0.4 mL/min
2009
23
11
Etienne Cavalier
HPLC
Wavelength: 254 nm
Mobile phase: Distilled water and acetonitrile (95:5 v/v) adjusted to pH3.0 with orthophosphoric acid.
Linearity: 6.47–1295 μg/ml for serum, 52.0–4144 μg/ml for urine sample
2008
19
12
Soo-Youn Lee
HPLC-MS/MS
Mobile phases: Water containing 2mM ammonium acetate and 0.1% formic acid (A) and methanol containing 2mM ammonium acetate and 0.1% formic acid (B).
Linearity: 0.5 and 1500 g/mL
2006
13
13
Domenic A. Sica
HPLC–UV
Wavelength: 254nm
Mobile phase: Aq. trifluoroacetic acid (0.1% TFA in deionized water (pH 2.2), v/v) and methanol gradient.
2005
20
CONCLUSION:
The accurate quantification of iohexol in pharmaceutical preparations is essential for ensuring product quality, efficacy, and patient safety. This review highlights various analytical techniques employed for iohexol qualification, including HPLC, UPLC, spectrophotometry, and advanced hyphenated methods such as LC-MS and LC-MS/MS. Among these, HPLC remains the most widely used due to its high sensitivity, robustness, and regulatory acceptance. However, emerging techniques like LC-MS provide superior specificity and lower detection limits, making them valuable for complex sample matrices. Future advancements in analytical methodologies, including automation, miniaturization, and greener chemistry approaches, are expected to enhance the efficiency and sustainability of iohexol analysis. Further research should focus on developing more rapid, cost-effective, and environmentally friendly analytical techniques to support quality control and regulatory compliance in the pharmaceutical industry.
CONFLICT OF INTREST:
The authors declare no conflicts of interest regarding the publication of this review article.
ACKNOWLEDGEMENTS:
We extend our heartfelt gratitude to Dr. Dinesh Chaple, Principal of Priyadarshini J. L. College of Pharmacy, Nagpur, for his invaluable guidance and unwavering support, which played a pivotal role in shaping the development of this review article.
REFERENCES
Wang, Z. et al. Degradation of iohexol by UV/chlorine process and formation of iodinated trihalomethanes during post-chlorination. Chem. Eng. J. 283, 1090–1096 (2016).
Braselton, W. E., Stuart, K. J. & Kruger, J. M. Measurement of serum iohexol by determination of iodine with inductively coupled plasma–atomic emission spectroscopy. Clin. Chem. 43, 1429–1435 (1997).
Hu, N. Comparison of separate measurement procedures for determination of GFR in chronic kidney disease: iohexol quantification by HPLC and UPLC. (2023).
Schulz, A. et al. A highly sensitive method for quantification of iohexol. Anal Methods 6, 3706–3712 (2014).
Ion, V. et al. Determination of iohexol by capillary blood microsampling and UHPLC-MS/MS. J. Pharm. Anal. 9, 259–265 (2019).
Wharton, T. & Wilson, L. J. Highly-Iodinated Fullerene as a Contrast Agent For X-ray Imaging. Bioorg. Med. Chem. 10, 3545–3554 (2002).
Salazar, M. Chemistry Review: CMC Assessment Section for NDA 205-383 Oraltag™ (Iohexol for Oral Solution). The Division of Medical Imaging Products (2015).
Holmaas, L., Cervenka, J., Homestad, Ø., Patel, A. D., & Hussain, K. International Patent Application for WO 2005/003080 A1. World Intellectual Property Organization (2005)..
Haavaldsen, J. Iohexol. Introduction.
Liu, Y.-Z. et al. Degradation of iohexol by UV irradiation: kinetics, pathways and iodinated trihalomethanes formation during post-chlorination. Desalination Water Treat. 83, 56–65 (2017).
Holleran, J. L. et al. Quantitation of iohexol, a glomerular filtration marker, in human plasma by LC–MS/MS. J. Pharm. Biomed. Anal. 189, 113464 (2020).
Vicente, F. B., Vespa, G. K., Carrara, F., Gaspari, F. & Haymond, S. Determination of iohexol in human serum by a semi-automated liquid chromatography tandem mass spectrometry method. Clin. Biochem. 48, 679–685 (2015).
Lee, S.-Y., Chun, M.-R., Kim, D.-J. & Kim, J. W. Determination of iohexol clearance by high-performance liquid chromatography-tandem mass spectrometry (HPLC-MS/MS). J. Chromatogr. B 839, 124–129 (2006).
El Assri, S. et al. Iohexol assay for direct determination of glomerular filtration rate: optimization and development of an HPLC-UV method for measurement in serum and urine. Clin. Chim. Acta 508, 115–121 (2020).
Giannakis, S. et al. Iohexol degradation in wastewater and urine by UV-based Advanced Oxidation Processes (AOPs): Process modeling and by-products identification. J. Environ. Manage. 195, 174–185 (2017).
Schwertner, H. A. & Weld, K. J. High-Performance Liquid-Chromatographic Analysis of Plasma Iohexol Concentrations. J. Chromatogr. Sci. 53, 1475–1480 (2015).
Soman, R. S., Zahir, H. & Akhlaghi, F. Development and validation of an HPLC-UV method for determination of iohexol in human plasma. J. Chromatogr. B 816, 339–343 (2005).
Chaloemsuwiwattanakan, T. et al. Simple and fast analysis of iohexol in human serums using micro?hydrophilic interaction liquid chromatography with monolithic column. J. Sep. Sci. 39, 3521–3527 (2016).
Cavalier, E. et al. Performance of iohexol determination in serum and urine by HPLC: Validation, risk and uncertainty assessment. Clin. Chim. Acta 396, 80–85 (2008).
Farthing, D. et al. Simple HPLC–UV method for determination of iohexol, iothalamate, p-aminohippuric acid and n-acetyl-p-aminohippuric acid in human plasma and urine with ERPF, GFR and ERPF/GFR ratio determination using colorimetric analysis. J. Chromatogr. B 826, 267–272 (2005).
Marko Jovanovi?, Raki?, T., Tumpa, A. & Jan?i? Stojanovi?, B. Quality by Design approach in the development of hydrophilic interaction liquid chromatographic method for the analysis of iohexol and its impurities. J. Pharm. Biomed. Anal. 110, 42–48 (2015).
De Baere, S. et al. Quantitative determination of exo- and endo-iohexol in canine and feline samples using high performance liquid chromatography with ultraviolet detection. J. Pharm. Biomed. Anal. 61, 50–56 (2012).
Annesley, T. M. & Clayton, L. T. Ultraperformance Liquid Chromatography–Tandem Mass Spectrometry Assay for Iohexol in Human Serum. Clin. Chem. 55, 1196–1202 (2009).
Reference
Wang, Z. et al. Degradation of iohexol by UV/chlorine process and formation of iodinated trihalomethanes during post-chlorination. Chem. Eng. J. 283, 1090–1096 (2016).
Braselton, W. E., Stuart, K. J. & Kruger, J. M. Measurement of serum iohexol by determination of iodine with inductively coupled plasma–atomic emission spectroscopy. Clin. Chem. 43, 1429–1435 (1997).
Hu, N. Comparison of separate measurement procedures for determination of GFR in chronic kidney disease: iohexol quantification by HPLC and UPLC. (2023).
Schulz, A. et al. A highly sensitive method for quantification of iohexol. Anal Methods 6, 3706–3712 (2014).
Ion, V. et al. Determination of iohexol by capillary blood microsampling and UHPLC-MS/MS. J. Pharm. Anal. 9, 259–265 (2019).
Wharton, T. & Wilson, L. J. Highly-Iodinated Fullerene as a Contrast Agent For X-ray Imaging. Bioorg. Med. Chem. 10, 3545–3554 (2002).
Salazar, M. Chemistry Review: CMC Assessment Section for NDA 205-383 Oraltag™ (Iohexol for Oral Solution). The Division of Medical Imaging Products (2015).
Holmaas, L., Cervenka, J., Homestad, Ø., Patel, A. D., & Hussain, K. International Patent Application for WO 2005/003080 A1. World Intellectual Property Organization (2005)..
Haavaldsen, J. Iohexol. Introduction.
Liu, Y.-Z. et al. Degradation of iohexol by UV irradiation: kinetics, pathways and iodinated trihalomethanes formation during post-chlorination. Desalination Water Treat. 83, 56–65 (2017).
Holleran, J. L. et al. Quantitation of iohexol, a glomerular filtration marker, in human plasma by LC–MS/MS. J. Pharm. Biomed. Anal. 189, 113464 (2020).
Vicente, F. B., Vespa, G. K., Carrara, F., Gaspari, F. & Haymond, S. Determination of iohexol in human serum by a semi-automated liquid chromatography tandem mass spectrometry method. Clin. Biochem. 48, 679–685 (2015).
Lee, S.-Y., Chun, M.-R., Kim, D.-J. & Kim, J. W. Determination of iohexol clearance by high-performance liquid chromatography-tandem mass spectrometry (HPLC-MS/MS). J. Chromatogr. B 839, 124–129 (2006).
El Assri, S. et al. Iohexol assay for direct determination of glomerular filtration rate: optimization and development of an HPLC-UV method for measurement in serum and urine. Clin. Chim. Acta 508, 115–121 (2020).
Giannakis, S. et al. Iohexol degradation in wastewater and urine by UV-based Advanced Oxidation Processes (AOPs): Process modeling and by-products identification. J. Environ. Manage. 195, 174–185 (2017).
Schwertner, H. A. & Weld, K. J. High-Performance Liquid-Chromatographic Analysis of Plasma Iohexol Concentrations. J. Chromatogr. Sci. 53, 1475–1480 (2015).
Soman, R. S., Zahir, H. & Akhlaghi, F. Development and validation of an HPLC-UV method for determination of iohexol in human plasma. J. Chromatogr. B 816, 339–343 (2005).
Chaloemsuwiwattanakan, T. et al. Simple and fast analysis of iohexol in human serums using micro?hydrophilic interaction liquid chromatography with monolithic column. J. Sep. Sci. 39, 3521–3527 (2016).
Cavalier, E. et al. Performance of iohexol determination in serum and urine by HPLC: Validation, risk and uncertainty assessment. Clin. Chim. Acta 396, 80–85 (2008).
Farthing, D. et al. Simple HPLC–UV method for determination of iohexol, iothalamate, p-aminohippuric acid and n-acetyl-p-aminohippuric acid in human plasma and urine with ERPF, GFR and ERPF/GFR ratio determination using colorimetric analysis. J. Chromatogr. B 826, 267–272 (2005).
Marko Jovanovi?, Raki?, T., Tumpa, A. & Jan?i? Stojanovi?, B. Quality by Design approach in the development of hydrophilic interaction liquid chromatographic method for the analysis of iohexol and its impurities. J. Pharm. Biomed. Anal. 110, 42–48 (2015).
De Baere, S. et al. Quantitative determination of exo- and endo-iohexol in canine and feline samples using high performance liquid chromatography with ultraviolet detection. J. Pharm. Biomed. Anal. 61, 50–56 (2012).
Annesley, T. M. & Clayton, L. T. Ultraperformance Liquid Chromatography–Tandem Mass Spectrometry Assay for Iohexol in Human Serum. Clin. Chem. 55, 1196–1202 (2009).
Vaibhavi Meshram
Corresponding author
Department of Pharmaceutical chemistry, Priyadarshini J. L. College of Pharmacy, Nagpur 440016, Maharashtra, India.


 10.5281/zenodo.15718168
10.5281/zenodo.15718168
