Shivajirao S. Jondhale College of Pharmacy, Asangaon, Thane, Maharashtra, India 421601.
The blood–brain barrier (BBB) is a highly specialised and dynamic interface that regulates the exchange of molecules between the systemic circulation and the central nervous system (CNS). It maintains brain homeostasis while restricting the entry of neurotoxic compounds. This selective permeability is primarily governed by endothelial tight junctions and a variety of transporter proteins that mediate influx and efflux of endogenous and exogenous substances. Influx transporters, such as glucose, amino acid, monocarboxylate, and nucleoside transporters, ensure the deliveryof nutrients and metabolic balance, while efflux transporters, including P-glycoprotein (P-gp) and multidrug resistance proteins (MRPs), protect the brain by removing xenobiotics. However, these same mechanisms pose major challenges for CNS drug delivery, particularly in the treatment of neurological disorders such as Alzheimer’s disease, Parkinson’s disease, and epilepsy. Dysfunction of BBB transporters contributes to disease progression and drug resistance, highlighting the need for novel therapeutic strategies targeting transporter modulation and BBB permeability. Understanding BBB transporter biology is therefore critical for advancing neuropharmacology and improving CNS drug design.
The brain serves as the central organ responsible for managing and coordinating all bodily systems. Its parenchymal cells receive a continuous supply of oxygen and essential nutrients—primarily glucose and amino acids—through a highly intricate network of blood capillaries. The brain's microvascular network is estimated to extend approximately 600–700 kilometres in total length, while the collective endothelial surface area of its vessels—including arteries, arterioles, capillaries, venules, and veins— covers about 20 square meters. The idea of the blood–brain barrier (BBB) was first introduced in the late 19th century by Paul Ehrlich, who found that when trypan blue dye was injected into a rat's bloodstream, it stained peripheral organs but not the brain or spinal cord. Later, Goldman expanded on these findings by injecting the same dye directly into the cerebrospinal fluid (CSF). He observed that in this case, the dye stained only the central nervous system (CNS), leaving the rest of the body's tissues unaffected.[1,2]
Structure and Function of the Blood-Brain-Barrier (BBB)
Maintaining the stability of the extracellular environment in brain tissue, along with shielding it from harmful substances and fluctuations in blood composition, is essential for proper neuronal function. This regulation is ensured by a specialised structure that separates the blood from the brain tissue, known as the blood–brain barrier (BBB).[3] Definitive proof of a permeability barrier in the brain was provided in 1909 by Edwin Goldman, a South African–German scientist. He observed that when a dye was injected into a rat's bloodstream, it colored all body tissues except the brain and spinal cord.
Conversely, when the dye was introduced into the cerebral ventricles, staining occurred only in the central nervous system, leaving the rest of the body unaffected.[4]
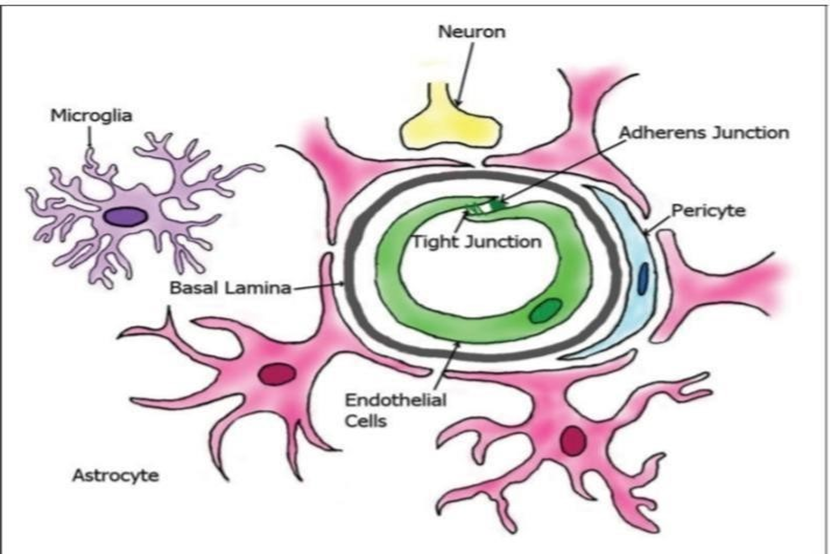
Fig.1 The Most Important Cellular Elements of BBB [5]
Anatomy and Physiology of the BBB
The blood–brain barrier (BBB) is a highly specialised physiological system composed mainly of brain capillary endothelial cells, basement membranes, pericytes, and glial cells such as astrocytes. The main role of the BBB is to protect the central nervous system (CNS) from harmful substances circulating in the external environment, while simultaneously preserving the homeostasis of the brain's internal environment.[6]
1. Endothelial Cells :
In contrast to blood vessels in the rest of the body, endothelial cells (ECs) of the BBB have a higher number of mitochondria, show very limited pinocytosis, and do not possess fenestrations.[7] The abundance of mitochondria in these cells is crucial for supporting various active transport processes, including the movement of ions, nutrients, and waste products into and out of the brain parenchyma. This enables tight control over the central nervous system (CNS) microenvironment, ensuring proper neuronal function. The high mitochondrial content in cerebrovascular endothelial cells may also explain the blood–brain barrier's vulnerability to oxidative stress. In addition, the normal and pathological functioning of endothelial cells is closely tied to mitochondrial health, and mitochondria are recognised as a significant contributor to brain disease.[8]
2. Tight Junctions :
Although breaking down adheres junctions (AJs) can increase BBB permeability, it is the tight junctions that (TJs) These play the main role in controlling paracellular transport across the barrier.[9] Tight junctions (TJs) constitute the main structural barrier of the BBB, strongly limiting the paracellular passage of both endogenous and exogenous substances that could be harmful to the brain. These junctions also generate a high trans-endothelial electrical resistance (TEER) of approximately 1500–2000 Ω·cm², which restricts the unrestricted movement of ions and solutes across the barrier. Tight junctions (TJs) are highly dynamic structures composed of several proteins, including junctional adhesion molecules (JAMs), occludin, claudins (such as claudin-1, -3, and 5), and membrane-associated guanylate kinase (MAGUK)-like proteins, like ZO-1, ZO-2, and ZO-3.[10]
3. Astrocytes :
Astrocytes play a crucial role in maintaining CNS homeostasis, providing protection, and supporting tissue repair. Impairment of astrocyte function or abnormal astrocyte activity is linked to brain ageing and the development of neurodegenerative disorders. These alterations in astrocytes are highly variable and often specific to particular brain regions.[11]
4. Pericytes :
Pericytes, which are integral to the neurovascular unit, are positioned between endothelial cells and astrocyte endfeet (Figure 1). They reside within the basement membrane that encases the endothelial cells, keeping them physically separate from both the endothelial cells and the astrocyte endfeet.[12] Existing studies report conflicting data regarding the proportion of the abluminal plasma membrane of endothelial cells that is covered by pericytes, with values ranging widely from 22% to 99% [13]
5. Basement Membrane :
The basement membrane of brain capillaries is a well-organised protein layer, approximately 50–100 nm thick, that envelops both endothelial cells and pericytes. It is a highly dynamic component of the BBB and is crucial for maintaining its structural and functional integrity. [14]The basement membrane is produced by both endothelial cells and pericytes, and its primary components include laminins, type IV collagen isoforms, fibrillins, vitronectin, fibronectin, elastin, nidogens, and heparan sulfate.[15]
6. Microglia :
Microglia are long-lived immune cells that reside within the CNS, making up approximately 12–16% of the brain’s total cell population. They serve as the primary defenders of the brain against immunerelated threats and play a key role in maintaining neuronal homeostasis.[16] Disruption of the BBB can influence microglial activity via interactions with activated endothelial cells, even without underlying neurodegeneration. Consequently, brain regions containing enlarged or activated microglia near blood vessels may indicate vascular injury and BBB dysfunction.[17]
Functions of the BBB
At the BBB, key membrane-bound proteins known as BBB trans porters, highly expressed in BECs, play a central role in selectively allowing the uptake of essential circulating substances while restricting the permeability of unwanted substances.[18]Transporter proteins are found in many tissues, especially within the epithelial and endothelial cells of organs that serve barrier functions, including the brain, placenta, and kidneys.[19]
The blood–brain barrier (BBB) is a physiological mechanism that regulates the permeability of cerebral capillaries, blocking certain substances, such as some drugs, from entering brain tissue while permitting others to pass freely. Its primary function is to protect the brain from fluctuations in the levels of blood ions, amino acids, peptides, and other components.[20]
Classification of BBB Transporters
1. Influx Transporters
Glucose Transporters
Approximately 20% of ingested D-glucose is utilised by the human brain. To reach the brain interstitial fluid or ventricles, D-glucose must cross the blood–brain barrier (BBB), which includes the barrier between the choroid plexus and cerebrospinal fluid (CSF) in the ventricles, the barrier separating the brain interstitium from the ventricles, and the barrier between circumventricular organs (CVOs) and the ventricles.[21]
Roles of glucose transporters during AD pathogenesis
The reduced expression of GLUT1/Glut1 and GLUT3/Glut3 proteins in Alzheimer’s disease may either act as an early concurrent event that worsens disease progression or represent an initial step in the causal pathway of AD pathogenesis.[22]
Diabetes: Glucose Transporters
Diabetes is a metabolic disorder marked by elevated blood glucose levels due to impaired insulin production, insulin action, or both. Individuals with type 1 (insulin-dependent) or type 2 (insulin-independent) diabetes frequently develop secondary complications, primarily affecting organs that take up glucose independently of insulin. The underlying pathophysiological mechanisms of these complications are largely linked to increased sorbitol accumulation, oxidative and nitrosative stress, depletion of endogenous antioxidants, enhanced lipid peroxidation, metabolic disturbances, and altered hormonal responses.[23]
Alzheimer’s Disease: Glucose Transporters in the Brain
The transport of glucose across the blood–brain barrier and neuronal plasma membranes can become a limiting factor when the brain experiences reduced energy availability or in pathological conditions such as Alzheimer's disease (AD), epilepsy, dementia, ischemia, and traumatic brain injury. From a histopathological perspective, AD is characterised by the accumulation of extracellular amyloid plaques (APs) and intracellular neurofibrillary tangles (NFTs) in the brain.[24]
Amino Acid Transporters
Broadly, amino acid transporters can be categorised based on their localisation in the luminal membrane, the abluminal membrane, or both. Functionally, these transporters operate through different mechanisms: antiporters facilitate the exchange of certain amino acids for others across the membrane, while symporters actively co-transport amino acids along with ions, following the ions' electrochemical gradient.[25]
Facilitative Amino Acid Transporters: Two sodium-independent transport systems, LAT1 or L, and y, mediate the facilitated exchange of large neutral AAs (both systems) and basic AAs (y ) in both luminal and abluminal membranes of ECs. They play a key role in delivering dietary essential neutral and basic AAs that cannot be synthesised within the brain[26]
Carrier-mediated transport of amino acid-like drugs: Essential amino acids must be transported from the blood into the brain, and specific transporters responsible for this process have been identified. In general, amino acid transporters are categorised based on their functional properties, including whether they depend on sodium and their substrate specificity.[27]
Monocarboxylic Acid Transporters
Proton-coupled monocarboxylate transporters (MCTs) were initially identified through their role in transporting lactate and pyruvate into human red blood cells, a process that can be markedly inhibited by α-cyano-4-hydroxycinnamate (CHC).[28]
Function of Monocarboxylate Acid Transporters:
Lactate transport across the plasma membrane is crucial during hypoxia, when glycolysis becomes the primary source of energy, and in tissues that depend on glycolysis to satisfy their normal energy requirements. In hypoxic conditions, glycolysis produces lactate, which must be removed from the cell to allow glycolysis to continue efficiently.[29]
Nucleoside Transporters
The first human nucleoside transporter (hNT) protein was discovered and cloned over two decades ago, and within a few years, the complete set of hNTs was fully characterised.[30]
Nucleoside transporters (NTs) are divided into two distinct families: the equilibrative nucleoside transporters (ENTs, SLC29), which mediate passive transport, and the concentrative nucleoside transporters (CNTs, SLC28), which function as active transport systems.[31] Nucleoside transporters (NTs) play a key role in the transmembrane movement of nucleosides. Nucleosides are derived from nucleotides, which are organic molecules composed of a nitrogenous base (either a purine or pyrimidine), a pentose sugar (ribose or deoxyribose), and one or more phosphate groups. Unlike nucleotides, nucleosides consist only of the sugar and base, lacking the phosphate component. The first NT family members were molecularly identified in human cells less than ten years ago, and since that time, these transporters have been found in most eukaryotic tissues.[32]
2. Efflux Transporters (Brain to Blood )
P-glycoprotein Transporters
Membrane transport proteins play a vital role in biology by eliminating toxic compounds from the cytosol and enabling the uptake of essential nutrients. This protective activity is critical for the survival of living organisms. Among these proteins, multidrug transporters are a group of transmembrane glycoproteins that actively move small lipophilic molecules out of the cell against their concentration gradients.[33]
Structure of P-glycoprotein
Embedded within the phospholipid bilayer, the P-glycoprotein (PGP) pump consists of two homologous transmembrane domains (TMDs), each made up of six α-helices that span the membrane and a cytoplasmic nucleotide-binding domain (NBD).[34]
PGP alterations in Disease:
Given that numerous factors can influence both the activity and expression of P-glycoprotein (PGP), it is unsurprising that PGP is implicated in various diseases. The protein p53, commonly referred to as "the guardian of the genome," has strong tumour-suppressive properties. It regulates processes such as cell-cycle arrest, apoptosis, senescence, and autophagy in response to cellular stress, DNA damage, oncogene activation, or hypoxic conditions.[35]
Multidrug Resistance Protein
Another protein commonly found in many multidrug-resistant (MDR) tumours is MRP1. Similar to P-glycoprotein, MRP1 belongs to the ABC transporter superfamily and is present in numerous MDR cell lines that lack P-gp expression. To date, seven MRP homologues have been identified in humans.[36]
Multidrug resistance (MDR) was initially identified in mammalian tumour cells, where it provides resistance to multiple chemotherapeutic drugs.[37] The protein encoded by these genes is P-glycoprotein (Pgp), a 170 kDa transporter capable of moving a wide range of substrates. Humans have two identified multidrug resistance proteins (P-glycoproteins, Pgps). MDR1 Pgp is encoded by the MDR1 gene in humans and by the Mdr1a and Mdr1b genes in rodents. This protein mediates the efflux of a variety of amphipathic and hydrophobic compounds, including drugs, from tumour cells. Under normal physiological conditions, MDR1 Pgp functions as an efflux pump in organs involved in the elimination of endogenous and xenobiotic substances, such as the liver and kidneys, as well as in tissues that protect against xenobiotic entry, including the small intestine, testes, placenta, and blood–brain barrier (BBB).[38]
Roles And Challenges in Neuropharmacology
Alzheimer’s Disease
Alzheimer's disease (AD) is a widespread neurodegenerative condition marked by a gradual loss of cognitive abilities along with behavioural and neuropsychiatric symptoms. It affects more than 55 million people globally, and the number of individuals living with AD is projected to rise substantially in the future. Alzheimer's disease is characterised by key pathological features such as the accumulation of amyloid-β plaques, abnormal tau phosphorylation forming neurofibrillary tangles, and the progressive loss of neurons and synapses. Together, these changes drive memory and cognitive decline, reduce patients' capacity to manage everyday tasks, and create substantial emotional and practical challenges for both individuals affected and their families.[39]
Current medications for Alzheimer’s disease, including cholinesterase inhibitors and NMDA receptor antagonists, mainly aim to relieve symptoms rather than halt disease progression. Although newer disease-modifying approaches like amyloid-β monoclonal antibodies offer potential benefits, their long-term effectiveness and safety still need further confirmation.[40]
Drug Development
The search for effective treatments for Alzheimer’s disease is a major priority in medical research worldwide. As of 2024, the U.S. Food and Drug Administration (FDA) has approved 12 drugs for AD treatment, while an additional 127 drugs are currently under investigation across 164 clinical trials. Numerous AD drugs are in clinical development, including phase III, II, and I trials. Recently approved drugs such as lecanemab and donanemab represent progress in treatment, but they mainly slow disease progression rather than cure it and may cause side effects like gastrointestinal discomfort, nausea, vomiting, and diarrhoea.[41]
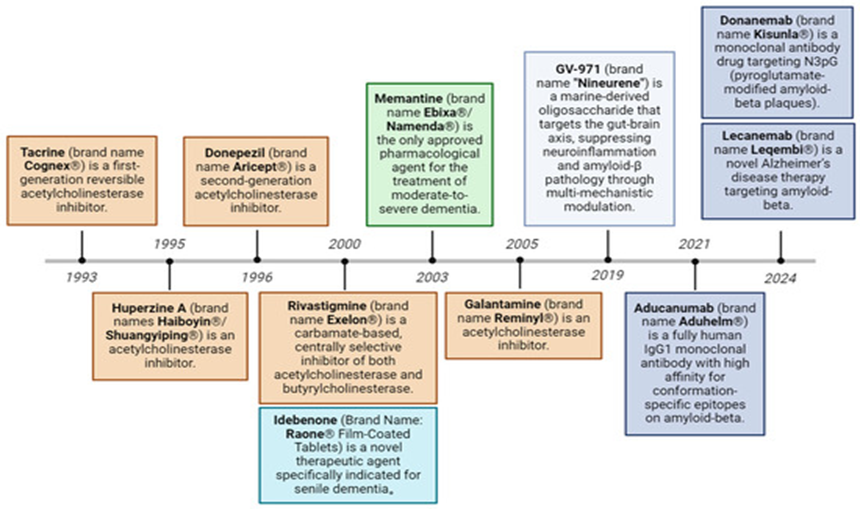
Fig.2 Evolution of approved therapeutic drugs for Alzheimer’s disease.
Current therapeutic strategies include acetylcholinesterase inhibitors, NMDA receptor antagonists, amyloid-targeting monoclonal antibodies, reactive oxygen species modulators, and gut-brain axis-targeting modulators.[41]
Research and Development of AD Drugs Targeting Aβ
Recent research indicates that therapies targeting amyloid-β can lower brain plaque levels and slow the progression of Alzheimer’s disease. Both the benefits and shortcomings of these approaches offer important lessons for guiding the development of more effective future treatments.
Lecanemab, a humanised IgG1 monoclonal antibody approved in January 2024, selectively targets soluble amyloid-β aggregates and, to a lesser extent, fibrillar amyloid. Its efficacy was demonstrated in the 18-month CLARITY AD trial involving patients with mild cognitive impairment or mild dementia and confirmed amyloid deposition. Weekly intravenous treatment with lecanemab significantly reduced brain amyloid plaques and slowed cognitive and functional decline, with a 27% reduction in decline on the CDR-SB and a 37% slowing on the ADCS-MCI-ADL compared with placebo.[42]
Donanemab, approved in December 2024, is a monoclonal antibody that targets insoluble, N-terminal truncated amyloid-β species found specifically within cerebral plaques. By binding to these forms of Aβ, it enhances plaque clearance through microglia-mediated phagocytosis. Results from the phase 3 TRAILBLAZER-ALZ trial demonstrated significant clinical benefits at 76 weeks, supporting its effectiveness in patients with early symptomatic Alzheimer’s disease who have confirmed amyloid and tau pathology.[43]
Challenges in AD Drug Development
Despite progress in Alzheimer’s drug development, few therapies reach clinical use with strong efficacy. This is largely due to the disease’s complex and multifactorial pathogenesis, involving multiple hypotheses such as amyloid-β, tau, cholinergic, vascular, and inflammatory mechanisms. As a result, treatments mainly provide symptomatic relief, and the lack of clearly defined targets contributes to frequent drug failure in clinical trials. Alzheimer’s disease shows significant genetic and environmental heterogeneity. Most cases are sporadic and result from complex interactions among multiple genes, environmental factors, and lifestyle influences.[44] Current FDA-approved drugs for Alzheimer's mainly relieve symptoms or slow progression, but cannot cure the disease. True radical treatments, like gene editing or nerve regeneration to address underlying causes, are still experimental and far from clinical use.
Therapies for the Prevention of AD
1. Pharmacological Treatment
Alzheimer’s disease, an age-related neurodegenerative disorder, demands accurate and early diagnosis, along with targeted treatment based on its underlying causes. Current therapies mainly aim to ease symptoms and slow disease progression, but they do not significantly reverse it. Therefore, focusing on prevention is considered a more effective approach to addressing this public health challenge.[45]
1.1 Symptomatic Treatment
Acetylcholinesterase Inhibitors: Acetylcholine (ACh) is essential for learning and memory. Research suggests that Aβ directly affects cholinergic systems, disrupting a negative feedback mechanism that normally regulates Aβ production. This disruption, along with abnormal Aβ buildup, decreases the efficiency of cholinergic signalling, particularly at alpha-7 nicotinic acetylcholine receptors. Several compounds, including tacrine, donepezil, rivastigmine, and galantamine, have been studied for Alzheimer’s treatment. Among them, rivastigmine, donepezil, and galantamine are approved drugs that boost acetylcholine levels and enhance cholinergic function by inhibiting acetylcholinesterase.[46]
N-Methyl-D-aspartate Receptor (NMDA) Antagonist
Excessive glutamate activity can cause calcium overload, mitochondrial dysfunction, and increased nitric oxide, leading to oxidative stress and neuronal cell death. NMDA receptor antagonists, like memantine, can prevent this overstimulation. Approved by the FDA in 2003, memantine is used for moderate-to-severe Alzheimer’s and offers modest cognitive benefits in mild-to-moderate cases.[47]
Other Neurotransmitter Systems
Muscarinic and nicotinic acetylcholine receptors are potential targets for Alzheimer’s treatment, but achieving selective activation has been challenging in clinical trials. EVP-6124 is currently being tested in a phase II trial.
Building on the cholinergic and NMDA-glutamate hypotheses, other neurotransmitter systems, especially in the hippocampus, are being explored for Alzheimer’s treatment. Serotonin receptors, present in brain regions linked to learning and memory, can influence acetylcholine release. In particular, blocking 5-HT6 receptors has been shown to increase acetylcholine, and several compounds targeting this mechanism are under clinical investigation for mild-to-moderate AD.[48]
1.2 Aetiology-Based Treatment
Amyloid Binders
Aβ accumulation in Alzheimer’s disease depends on its concentration, resulting from increased amyloidogenic processing of APP and impaired peptide clearance. Reduced activity of Aβ-degrading enzymes like neprilysin and insulin-degrading enzyme, along with the influence of ApoE, supports the view of AD as a metabolic disorder. Targeting the formation of extracellular Aβ plaques is a strategy for disease-modifying Alzheimer’s treatments. Although Aβ biomarkers can correlate with cognitive decline before plaque formation, inhibitors of Aβ aggregation have progressed to clinical trials.[49]
Tau Therapies
Therapies also aim to prevent the aggregation of hyperphosphorylated tau into neurofibrillary tangles. Immunotherapy approaches include AADvac1, the first tau-targeting vaccine tested in clinical trials, and ACI-35, a liposomal-based vaccine currently under investigation. Tau phosphorylation inhibitors like tideglusib (a GSK-3β inhibitor) have shown no significant benefits, while CDK5 is being explored as another potential drug target.[50]
Anti-Aβ Aggregation Compounds
Recent research has focused on preventing Aβ formation or aggregation. Small-molecule inhibitors in clinical trials—such as tramiprosate, clioquinol, scylloinositol, and epigallocatechin-3-gallate—can stabilise Aβ monomers but have notable side effects. Additionally, synthetic β-sheet breaker peptides like azetidine-2-carboxylic acid and its derivatives reduce Aβ-induced cell damage by preventing fibril formation and have shown improvements in spatial memory. Stemazole has been found to protect SH-SY5Y cells from Aβ-induced toxicity by reducing Aβ aggregation. Similarly, compounds like curcumin, T718MA, and SK-PC-B70M offer neuronal protection against Aβ-related damage.[51]
2. Non-Pharmacological Treatments
Nonpharmacological approaches play a key role in preventing Alzheimer's disease or supporting other treatments. Prevention strategies are generally categorised into lifestyle-based measures and those involving diet or chemical compounds.
2.1 Lifestyle
Lifestyle-based prevention of Alzheimer’s includes physical exercise, mental stimulation, calorie restriction, and social engagement. While aerobic exercise was linked to reduced AD-related deficits in large cohort studies, results were inconsistent in studies with smaller sample sizes. Exercise has been shown to promote hippocampal neurogenesis and improve learning in ageing rodents. Its neuroprotective effects are thought to occur through three main mechanisms: (1) release of neurotrophic factors like BDNF, IGF-1, NGF, and VEGF, which enhance neurogenesis and synaptic plasticity via CREB activation; (2) reduction of oxidative stress in the hippocampus, including increased superoxide dismutase and endothelial nitric oxide synthase; and (3) peripheral signals, such as BDNF and energy restriction, that support active neuronal networks.[52]
2.2 Diet and Chemical Substances
Dietary supplements, including vitamins B6, B12, folate, and vitamins C, D, and E, have been studied for Alzheimer's prevention. Research on B vitamins has shown mixed results: a two-year study found that supplementation reduced whole-brain atrophy compared to placebo, while other studies reported different outcomes. Folic acid may offer neuroprotection by epigenetically reducing amyloid-β accumulation. However, studies with vitamin E, alone or combined with vitamin C, showed no protective effect against Alzheimer’s over three years, while vitamin D supplementation has been found to enhance cognitive performance.[53]
Parkinson’s Disease
Parkinson’s disease (PD) is a neurodegenerative disorder that primarily impacts the basal ganglia, leading to difficulties in controlling movement. It is the second most prevalent neurodegenerative condition after Alzheimer’s disease. PD manifests through symptoms such as slowed movements (bradykinesia), altered posture, abnormal walking patterns, and shorter steps. Additionally, individuals with PD often experience vocal problems, psychological issues, and reduced facial expressions, all of which can significantly affect their quality of life.[54]
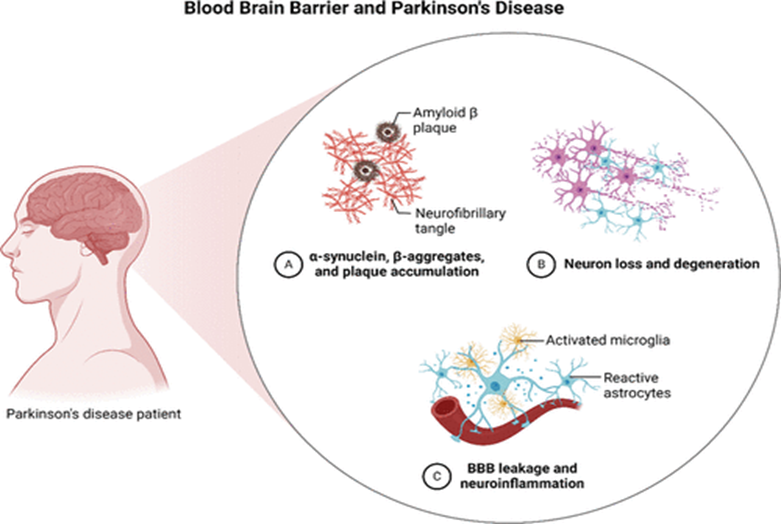
Fig.3 Schematic diagram of the blood–brain barrier (BBB) and its function in Parkinson’s disease.
The diagram illustrates the BBB (A) and its impairment in Parkinson’s disease (B), resulting in the buildup of harmful proteins such as amyloid-β plaques, neurofibrillary tangles, α-synuclein, and β-aggregates.
Significance of Blood Barriers in Parkinson’s Disease
Role of Blood–Brain Barrier Impairment
The blood–brain barrier (BBB) is a protective barrier separating the brain from the bloodstream, which contains nutrients, pathogens, and potentially harmful substances. It helps maintain brain homeostasis and shields the brain from toxins by selectively permitting only certain molecules to pass. Specifically, the BBB is selectively permeable, allowing only lipid-soluble molecules with a size of 400–600 Da to cross.[55]
Role of Blood–Retina Barrier Impairment
Visual impairments in Parkinson's disease are closely linked to the blood–retina barrier (BRB), which controls the exchange of substances between the bloodstream and the retina. The BRB consists of two parts: an inner barrier, similar to the blood–brain barrier (BBB), located in the retinal microvasculature, and an outer barrier at the retinal pigment epithelial layer that facilitates nutrient and solute transport while protecting the retina from blood-borne toxins. Both the BBB and BRB are components of the neurovascular unit, helping maintain central nervous system function by regulating chemical transport and supporting stable cell interactions.[56]
Current treatment approaches for the PDs
Pharmacological treatments of PDs
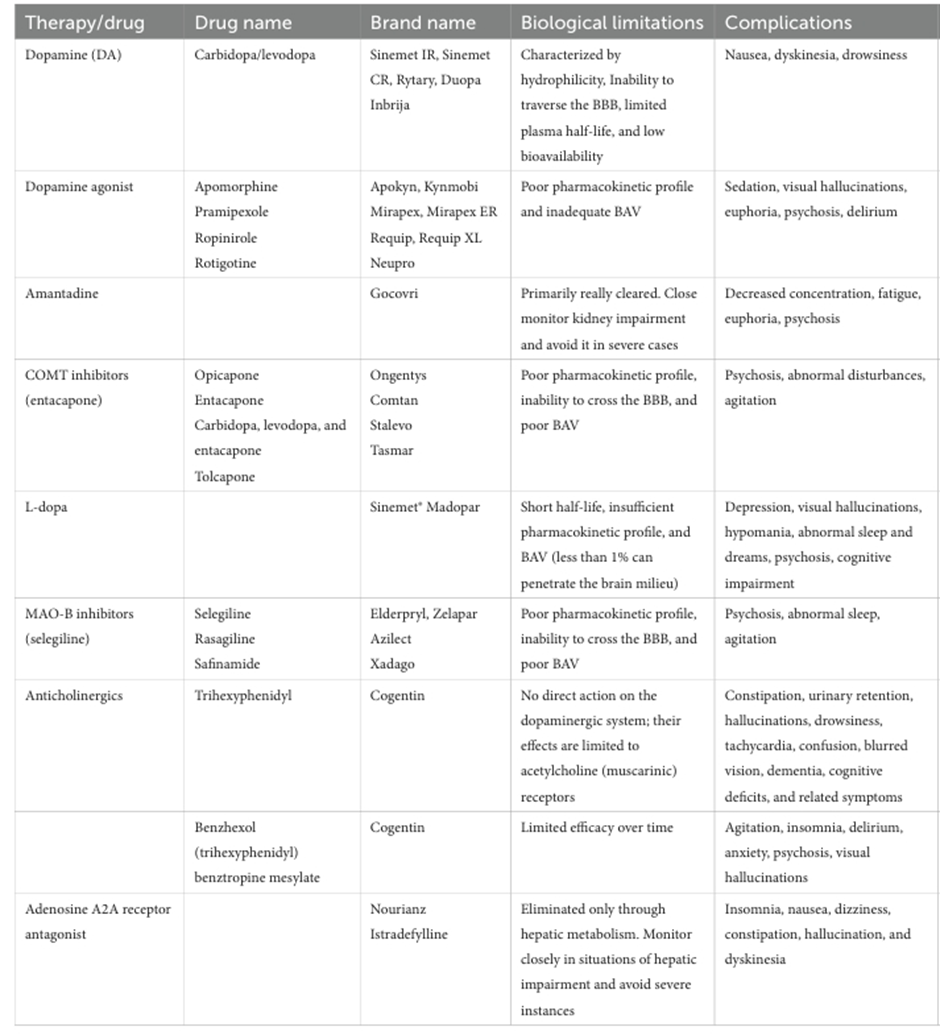
Pharmacological therapy for Parkinson’s disease includes agents such as levodopa combined with carbidopa, dopamine agonists, MAO-B inhibitors, COMT inhibitors, anticholinergic medications, and amantadine. Levodopa is the most effective and widely used treatment, serving as a dopamine precursor that crosses the blood–brain barrier and is then converted into dopamine within the brain. Carbidopa is co-administered to prevent the premature conversion of levodopa to dopamine outside the central nervous system, which helps reduce side effects such as nausea and allows more levodopa to reach the brain.[57]
Table.1 Biological limitations and complications associated with the current treatment Approaches for PD.[58]
Drug Therapy for Parkinson’s Disease
Although new treatment developments are expected, current management strategies for Parkinson’s disease are largely focused on symptomatic drug therapy that targets neurotransmitter regulation in the brain. The hallmark pathological feature of PD is the degeneration of nigrostriatal dopaminergic neurons, making dopamine replacement therapy the cornerstone and most essential treatment approach for patients.
In the first part of this review, we discuss the medications currently available for the symptomatic management of Parkinson’s disease. As understanding of the disease’s pathophysiology, including the mechanisms underlying neuronal damage, continues to improve, efforts are underway to develop therapies that can modify disease progression. The second part of the review, therefore, focuses on potential disease-modifying agents that are presently in phase II or more advanced stages of clinical trials.
I. Symptomatic Therapy
Dopamine deficiency in the striatum of patients with Parkinson's disease was first identified in the 1960s, leading to the development of levodopa as a treatment that improves symptoms and extends patient survival. Levodopa remains the gold standard therapy for PD. However, by the mid-1970s, it was recognised that long-term use of levodopa can result in motor complications, including the wearing-off effect.[59]
1. Levodopa
1. Problems with current levodopa treatment: Levodopa is the most effective medication for improving motor symptoms in Parkinson’s disease. However, it has a short blood half-life of approximately 90 minutes, leading to fluctuations in drug levels that can cause variations in clinical response in advanced stages of the disease, known as the wearing-off phenomenon. As a result, alternative formulations or routes of administration aimed at extending levodopa’s duration of action are currently being explored.
2. Levodopa/carbidopa intestinal gel: To address the short half-life of levodopa and enhance motor control during daytime activities, levodopa/carbidopa intestinal gel (LCIG) was developed. This formulation is delivered as a continuous infusion into the upper jejunum via a gastrostomy tube during waking hours. LCIG was approved in Japan in 2016. Studies in East Asian patients who experienced about three hours of daily off-time showed that LCIG reduced off-time by approximately four to five hours and increased on-time without troublesome dyskinesia by five to six hours.[60]
3. Levodopa inhalant: An inhaled form of levodopa (CVT-301) has been approved in the United States, though not in Japan, for use as a rescue treatment during off-time. This formulation allows levodopa to be inhaled and quickly absorbed through the lungs. In a phase III trial using an 84-mg capsule containing 42 mg of levodopa, motor symptoms during off-time showed improvement within 10 minutes and were significantly better after 30 minutes compared with pre-inhalation levels.[61]
2. Monoamine oxidase-B inhibitors
1. Novel MAO-B inhibitors: In Japan, selegiline was approved in 1998 and has been in use for over 20 years, while newer MAO-B inhibitors, rasagiline and safinamide, were approved in 2018 and 2019, respectively. Rasagiline, when administered alone at 1 mg/day, improves motor symptoms in early-stage Parkinson’s disease. In advanced-stage patients experiencing motor complications while on oral levodopa, adding 0.5 or 1 mg/day of rasagiline significantly reduces off-time and enhances motor function.[62]
2) COMT inhibitors: Entacapone, a COMT inhibitor, enhances levodopa’s entry into the brain by preventing its peripheral metabolism by COMT. It has been used in Japan for many years, and opicapone was approved as a second COMT inhibitor in 2020. In Parkinson’s patients experiencing motor complications while on oral levodopa, opicapone at doses of 25 or 50 mg/day significantly reduced off-time and increased on-time without causing troublesome dyskinesia compared with placebo.[63]
II. Disease-modifying Therapy
Parkinson’s disease is a heterogeneous disorder, and its underlying pathophysiology is becoming better understood. Key factors include mitochondrial and lysosomal dysfunction, toxic α-synuclein aggregation, neuroinflammation, oxidative stress, and other cellular disturbances. These mechanisms represent potential targets for disease-modifying therapies aimed at addressing the root causes of the disease. Many studies are ongoing to identify drugs that can slow PD progression, and new compounds are currently in development.
1.α-synuclein targeting therapy
Immunisation for α-synuclein: α-Synuclein is a 140-amino-acid protein encoded by the SNCA (synuclein alpha) gene. While its normal physiological function is not fully understood, its aggregation is harmful to neurons. Research is ongoing into immunotherapy targeting α-synuclein oligomers. In passive immunisation, BIIB054 (cinpanemab), a monoclonal antibody that binds to oligomeric and fibrillar α-synuclein, demonstrated good safety and tolerability in a phase 1 trial. However, its development was discontinued in 2021 after the phase 2 SPARK study showed it did not improve motor symptoms. Another monoclonal antibody, PRX002 (prasinezumab), is also being investigated for its action against aggregated α-synuclein.[64]
2. Enhancers of β-glucocerebrosidase
Ambroxol: Ambroxol, an expectorant, has been found to enhance GBA enzyme activity in cells with GBA mutations and improve lysosomal function in cells from patients with GBA mutation–associated Parkinson’s disease. In the AiM-PD study, a non-randomised and uncontrolled trial, ambroxol treatment led to improvements in motor function in PD patients both with and without GBA-1 mutations.[65]
Venglustat (GZ/SAR402671): Venglustat is a glucocerebroside synthase inhibitor intended to lower glucosylceramide production. This "substrate reduction therapy" targets an upstream enzyme to decrease the accumulation of harmful substrates and was expected to benefit Parkinson’s patients with GBA mutations. However, the phase 2 MOVES-PD trial evaluating venglustat in PD patients with GBA mutations failed to achieve its primary or secondary endpoints, leading to the discontinuation of further follow-up in 2021.
3. Medication with neuroprotective effects
c-Abl inhibitor: Abelson (c-Abl) is a non-receptor tyrosine kinase that becomes activated in response to oxidative and cellular stress. It contributes to Parkinson’s disease pathology by promoting α-synuclein aggregation and Lewy body formation, impairing autophagy, causing mitochondrial dysfunction, and activating microglia. Thus, targeting c-Abl could potentially affect the underlying mechanisms of Parkinson’s disease. Certain c-Abl inhibitors, already approved for treating chronic myelogenous leukaemia, have shown neuroprotective effects in recent studies using PD model mice.[66]
Epilepsy
Epilepsy refers to a set of long-term, non-communicable neurological conditions defined by recurrent, unprovoked seizures caused by abnormal electrical activity in the brain. The development of epilepsy in a previously healthy brain is known as epileptogenesis. Epilepsy can arise from various causes, including head trauma, brain tumours, infections such as meningitis or encephalitis, stroke, congenital abnormalities, and, in some cases, imbalances in substances like blood sugar or sodium levels. Epilepsy is the third most prevalent long-term neurological condition and is associated with reduced life expectancy. It affects about 50 million people worldwide, roughly 1% of the global population, with nearly 80% of those affected living in low- and middle-income countries.[67]
Roles in Epilepsy
A. Role of Anti-seizure Medications(ASMS)
The Primary pharmaceutical role in epilepsy is seizure suppression. ASMS act by modulating neuronal excitability through different mechanisms, including :
1. Modulation of Voltage-Gated Ion Channels :
Voltage-gated ion channels are membrane-spanning proteins that regulate ion movement in response to changes in membrane potential, enabling the initiation and transmission of action potentials. They are selective for specific ions, including sodium, calcium, potassium, and chloride, and share similar activation mechanisms. Because of their essential role in neuronal signalling, these channels are frequent targets of many antiseizure medications (ASMs).[68]
2. Modulation of Voltage-gated Sodium Channels :
Voltage-gated sodium channels (VGSCs) are membrane proteins that play a key role in controlling cellular electrical activity. They are primarily located in neurons, neuroendocrine cells, skeletal muscle, and cardiac tissue. Structurally, VGSCs are highly glycosylated complexes composed of one large α subunit (~260 kDa) and two smaller auxiliary β subunits (~35 kDa each).[69]
3. Modulation of Voltage-gated Calcium Channels :
Voltage-gated calcium channels (VGCCs) are membrane-spanning proteins that permit calcium ions to enter cells during membrane depolarisation, enhancing further depolarisation. The incoming calcium also serves as a second messenger, triggering multiple intracellular signalling pathways and downstream cellular responses.[70]
4. Enhancement of Inhibitory Neurotransmission :
Gamma-aminobutyric acid (GABA) is the primary inhibitory neurotransmitter in the vertebrate central nervous system, where it is essential for lowering neuronal excitability. It is produced from glutamate and broken down by GABA transaminase (GABA-T) through the GABA shunt, a metabolic pathway that bypasses two steps of the tricarboxylic acid (TCA) cycle.[71]
B. Disease Modification and Neuroprotection
Beyond symptomatic control, some ASMs demonstrate neuroprotective properties, including reduction of excitotoxicity, oxidative stress, and inflammation. Valproate and topiramate, for example, have been shown in experimental models to limit neuronal injury following seizures or brain insults, suggesting a potential role in modifying disease progression [72]
Development in the Anti-Seizure Medications :
Table 2. Overview of FDA-approved ASMs for epilepsy management.[73,74]
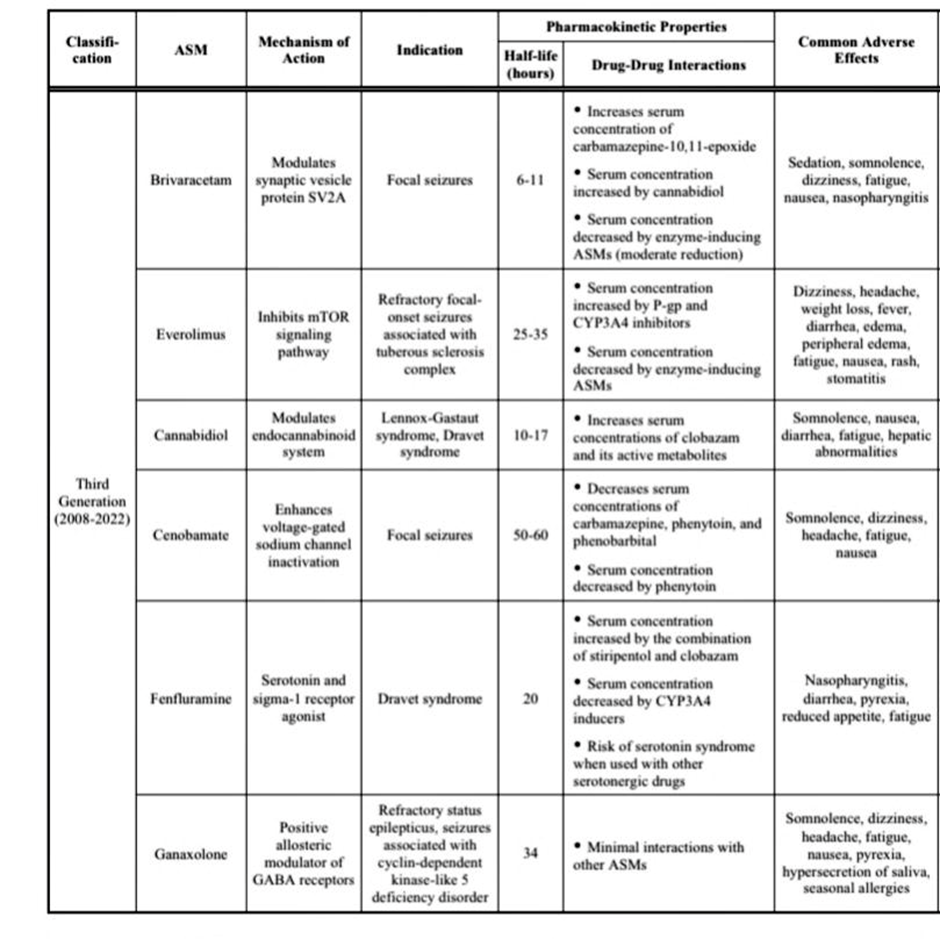
Cannabidiol (CBD), a cannabinoid obtained from the cannabis plant, has demonstrated effectiveness in treating seizures associated with Lennox–Gastaut and Dravet syndromes. It may also provide therapeutic benefits for both children and adults with severe, treatment-resistant epilepsy. Beyond epilepsy, research suggests that CBD could help alleviate symptoms of anxiety, pain, dystonia, Parkinson’s disease, Crohn’s disease, and other disorders. Cannabinoids have unique, multimodal analgesic properties, enabling them to influence multiple pain pathways. However, a key concern with CBD use is its potential to interact with other medications, including opioid painkillers, antidepressants, and other anti-seizure medicines.[75]
Cenobamate is a recently developed anti-seizure medication used for the treatment of focal-onset seizures in adults. As a carbamate derivative, it shares structural similarities with other alkyl-carbamate drugs such as felbamate and retigabine, which contribute to comparable mechanisms of action. These agents act as positive allosteric modulators of GABAA receptors, thereby strengthening inhibitory neurotransmission. In addition, cenobamate selectively inhibits voltage-gated sodium channels in their inactivated state, reducing the persistent sodium current rather than the transient current.[76]
CHALLENGES IN THE DEVELOPMENT OF ANTISEIZURE MEDICATIONS
Over time, various strategies have been developed to treat epilepsy, including medications, surgical interventions, therapeutic devices, and ketogenic diets. Despite these options, anti-seizure medications (ASMs) continue to be the primary treatment for most patients.
A. Drug-resistant Epilepsy: Drug-resistant epilepsy (DRE), also called refractory epilepsy, remains a major challenge, prompting the development of new treatments. According to the International League Against Epilepsy (ILAE), DRE is defined as the failure to achieve sustained seizure freedom after trying two properly selected and tolerated anti-seizure medication regimens, either alone or in combination. In simple terms, people with epilepsy whose seizures do not respond to anti-seizure medications are classified as having drug-resistant epilepsy (DRE). These patients are at higher risk of death and mental health issues compared to other individuals with epilepsy, highlighting the urgent need for more effective treatments for DRE.[77]
B. Adverse Effects of ASMs: Adverse effects are harmful or unexpected responses to a drug. Anti-seizure medications (ASMs) can cause such effects across multiple body systems, posing a significant challenge in their development. Since many epilepsy patients need long-term or lifelong treatment, experiencing adverse effects is common, making these reactions a major factor in treatment failure.
Anti-seizure medications (ASMs) primarily affect neuronal activity in the central nervous system, so most of their side effects are neurological. Commonly reported effects include sedation, dizziness, fatigue, headache, ataxia, blurred vision, tremors, cognitive impairment, and behavioural changes. These effects are often associated with the ability of ASMs to cross the blood-brain barrier, with BBB-penetrating drugs more likely to produce such unwanted reactions.[78]
Brain Tumour
The brain is the most sensitive organ in the human body, and its function can be impaired by various conditions, including encephalitis, neurological disorders, multiple sclerosis, stroke, and tumours. Developing new treatments for these diseases is extremely challenging, and effective therapies are lacking for most brain disorders. A major reason for the difficulty in developing drugs for brain diseases is the presence of the blood-brain barrier (BBB). Among brain disorders, brain tumours often have a poor prognosis, which depends on the tumour's type and grade. The BBB makes delivering drugs to brain tumours particularly difficult.
Brain tumours are broadly divided into two main types: primary tumours, which originate in the brain, and secondary tumours, which result from cancer cells that spread from other parts of the body. [79]
Current Roles of Pharmaceuticals in Brain Tumour Therapy
1. Conventional Chemotherapy
Traditional chemotherapeutics (e.g., temozolomide) act by interfering with DNA synthesis and tumour cell proliferation. Temozolomide remains a cornerstone in GBM treatment post-surgery and radiation, improving survival modestly, but tumour recurrence is frequent due to intrinsic and acquired resistance mechanisms.
2. Targeted Therapies
Targeted small-molecule inhibitors and agents against specific molecular drivers offer precision oncology opportunities. These are designed to exploit tumour-specific signalling pathways or mutations, but many fail to penetrate the brain in effective concentrations due to physiological barriers.
3. Immunotherapy and Combinatorial Strategies
Immunotherapies including immune checkpoint inhibitors and oncolytic virotherapy are being explored to harness the patient’s immune system against brain tumour cells. Additionally, combination therapies blending immunotherapy with conventional pharmaceuticals aim to overcome monotherapy limitations and target multiple tumour survival pathways simultaneously.[80,81]
Challenges in Brain Tumour Therapy
Treating brain tumours is much more challenging than treating peripheral tumours (Figure 1). Physiological barriers, including the blood-brain barrier (BBB) and blood-brain tumour barrier (BBTB), along with overactive efflux pumps, limit drug delivery to the central nervous system (CNS) and the tumour site. Additionally, the intrinsic features of brain tumours—such as infiltration, invasion, high heterogeneity, drug resistance, and immune evasion driven by the tumour microenvironment (TME) and cancer stem cells (CSC)—further reduce treatment effectiveness, resulting in high rates of treatment failure and tumour recurrence.
1. Blood-brain barrier and blood-brain tumour barrier
When a brain tumour grows beyond 2 mm³, angiogenesis disrupts the normal structure and function of the blood-brain barrier (BBB), leading to the formation of the blood-brain tumour barrier (BBTB). Although passive targeting through the enhanced permeability and retention (EPR) effect has been considered a key mechanism for nanoparticle accumulation, the small vascular pore size in brain tumours (only 7–100 nm) makes the EPR effect weak. As a result, delivering drugs to brain tumour sites via EPR remains challenging. Therefore, the BBTB is regarded as a major barrier to drug delivery, severely limiting therapeutic access to tumour tissue.[82]
2. Tumour Microenvironment
The tumour microenvironment (TME) is composed of tumour cells, cancer stem cells, blood and lymphatic vessels, immune cells, fibroblasts, and the extracellular matrix, creating a supportive setting for tumour growth, division, angiogenesis, and metastasis (Petrova et al., 2018). The TME protects tumour cells through several mechanisms. For example, elevated vascular endothelial growth factor (VEGF) activity promotes rapid microvessel proliferation. Tumour cells also use cytokines and growth factors from abnormal blood vessels to obtain nutrients, which in turn stimulates the proliferation and invasion of fibroblasts and macrophages, contributing to drug resistance.[83]
Drug Delivery Approaches and Current Advances in Brain Tumour Therapy
Most drugs for brain tumours are ineffective because they cannot efficiently cross the blood-brain barrier (BBB). Currently, researchers are focused on finding ways to overcome this challenge, and it is expected that improved drug delivery methods could greatly benefit many brain tumour patients.
1. Modification of Existing Drugs: A drug's ability to cross the blood-brain barrier (BBB) depends on several factors, including its molecular size (ideally under 500 Da), charge (low hydrogen bonding potential), and lipophilicity (should be high) [186]. Therefore, chemical modification of brain tumour drugs involves adjusting these properties to make the drug smaller, better balanced in charge, and more lipid-soluble.[84]
2. Nanosystem-Based Delivery: Nanosystems are colloidal carriers, primarily made up of liposomes and polymeric nanoparticles, though other types—such as solid lipid nanoparticles, polymeric micelles, and dendrimers—have also been explored recently. These nanosystems typically range in size from 1 to 1000 nm.[85]
3. Delivery Systems Used in Gene Therapy: Brain tumours can be effectively treated by directly implanting a therapeutic gene into the brain using a viral vector. This approach is highly targeted, as the implanted volume is very small (<1 mm³), resulting in localised expression of the introduced gene. However, modifications to the gene could lead to widespread expression throughout the entire brain tumour environment.[86]
Table 3: Recent Modification of a Few Important Brain Tumour Drugs
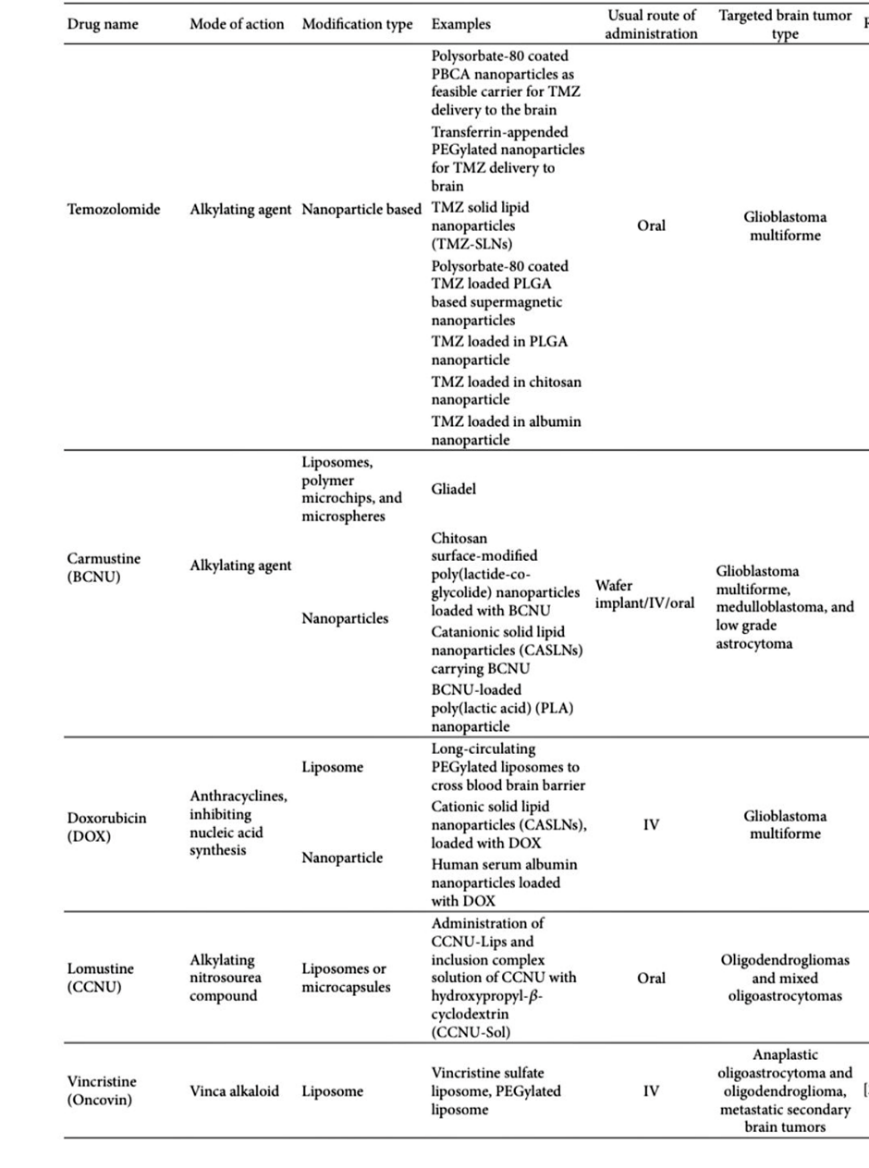
4. Effective Delivery of Therapeutic Peptides: To improve brain cancer therapy, selective peptides have recently been developed. The discovery of these novel peptides is facilitated by identifying specific protein or peptide receptors and tumour-associated proteins expressed in brain cancer cells. Small peptides offer several advantages over monoclonal antibodies (mAbs) and larger proteins, which are bulky, more toxic, and cross the blood-brain barrier (BBB) poorly. Peptides are beneficial because they can penetrate the BBB in brain tumours, are easier to synthesise and modify, and exhibit good biocompatibility. [87]
CONCLUSION
The blood-brain barrier (BBB) maintains brain homeostasis through specialized cells and transporter proteins. Influx transporters supply essential nutrients to the brain, while efflux transporters protect the CNS by removing toxins and drugs. Although crucial for normal function, these transporters hinder effective drug delivery to the brain. In disorders such as Alzheimer's disease, Parkinson's disease, epilepsy, and brain tumors, altered transporter activity contributes to disease progression and drug resistance. Current therapies are mainly symptomatic, but emerging strategies targeting BBB transporters and advanced drug delivery systems offer promise for improving CNS treatment outcomes.
REFERENCES






Neha Patil, Gauri Bhamare, Snehalata Mali, Kalyani Patil, Swapnil Pradhan, Raj Patil, BBB Transporters and Their Roles and Challenges in Drug Delivery System, Int. J. of Pharm. Sci., 2026, Vol 4, Issue 3, 1863-1886. https://doi.org/10.5281/zenodo.19072162
 10.5281/zenodo.19072162
10.5281/zenodo.19072162
