1 Department of Pharmacognosy and Natural Medicine, Faculty of Pharmacy, University of Uyo, Uyo, Nigeria.
2,4 Department of Pharmacognosy, Faculty of Pharmacy, University of Benin, Benin City, Nigeria.
3 Department of Pharmacology and Toxicology, Faculty of Pharmacy, University of Benin, Benin City, Nigeria.
Lonchocarpus griffonianus is traditionally used in southern Nigeria to treat ailments related to tumors, but its bioactive compounds remain underexplored. This study aimed to isolate, characterize, and evaluate the cytotoxic activity of secondary metabolites from the stem bark of L. griffonianus. Bioassay-guided fractionation was employed to isolate pure compounds, which were characterized using ¹H NMR, COSY, mass spectrometry, and melting point analysis. The cytotoxic activities of the isolated compounds were evaluated using the MTT assay against PC3 (prostate cancer), HeLa (cervical cancer), and BJ (normal fibroblast) cell lines. Spectroscopic analysis identified two compounds: lupeol (LO1) and ?-sitosterol (LO2). Lupeol showed characteristic NMR signals, including olefinic protons at ? 4.55 and ? 4.67. At the same time, ?-sitosterol displayed diagnostic signals, including a deshielded methine at ? 3.57 (C3) and an olefinic proton at ? 5.20 (C6). Mass spectra confirmed their molecular formulas as C??H??O (lupeol) and C??H??O (?-sitosterol). In cytotoxic assays, lupeol demonstrated higher inhibition against PC3 (40.8%) and HeLa (36.6%) compared to ?-sitosterol (23.6% and 25.1%, respectively), while showing lower toxicity to normal BJ cells (14.1%). Isolating lupeol and ?-sitosterol supports the traditional use of L. griffonianus as an anticancer remedy. Lupeol, in particular, exhibits selective toxicity to cancer cells and could be a promising lead for the development of new anticancer drugs.
Medicinal plants remain essential sources of bioactive compounds, especially in regions where traditional medicine is still a core part of primary healthcare 1. Ethnopharmacological evidence across multiple Lonchocarpus species reveals a diverse range of traditional uses, including treatments for tumors, headaches, skin diseases, malaria, diabetes, ulcers, and inflammatory conditions 2–4. Notably, L. araripensis has produced compounds with antinociceptive, anti-inflammatory, and gastroprotective properties 5. Phytochemical studies across the genus have revealed numerous polyphenolics, including aurones, chalcones, flavanones, rotenoids, and isoflavones 6, many of which exhibit significant pharmacological effects. Additionally, rotenoids such as rotenone and deguelin, found in species like L. urucu, act as powerful insecticidal and piscicidal agents, with increasing interest in their potential anticancer uses 7.
Despite the rich ethnobotanical and phytochemical background of the genus, L. griffonianus (Fabaceae) remains relatively unexplored. Ethnobotanical surveys indicate its use for treating tumor-related ailments in southern Nigeria, especially within the Oro community. Recent pharmacological tests using a methanol extract and its dichloromethane fraction have demonstrated strong cytotoxic and growth-inhibiting effects in tadpole and Sorghum bicolor radicle models 8, confirming its traditional use in the treatment of tumors. Additional studies have also verified its low toxicity in Sprague-Dawley rats, supporting its potential for safe medicinal use 9.
To date, the specific compounds responsible for the observed bioactivities have not been isolated or structurally identified. This creates a significant gap because linking pharmacological effects to specific chemical entities is essential for scientifically validating traditional claims and developing the plant into a phytotherapeutic agent. Therefore, this study aims to isolate and characterize the bioactive compounds in L. griffonianus stem bark that are responsible for the cytotoxic effects.
MATERIALS AND METHODS
Laboratory Materials and Equipment
Mass spectrometer (JEOL JMS 600H-1 model), nuclear magnetic resonance spectrometer (Bruker Avance 600 MHz model), thermoregulated water bath (WNB 22), TLC precoated plates (Merck), TLC tank, chromatography column, spray gun, oven, centrifuge (CF-30 model), mortar and pestle, TLC silica gel, silica gel (60-120 micron), and electric milling machine (Chris Norris, England).
Chemicals and reagents
chemicals used in this work include: n-hexane (BDH), dichloromethane (BDH), ethyl acetate (Lobachem), acetone (BDH), ethanol (BDH), methanol (Lobachem), toluene (BDH), DMSO (Gaylord), Tween-80, 10% formal-saline, concentrated sulfuric acid (Pharmatrend), gentian violet, hematoxylin and eosin, and the Accubind tPSA ELISA kit (USA), Accubind Estradiol ELISA kit (USA), and Accubind Testosterone ELISA kit (USA) P
Plant Collection, Identification, Extraction, and Partitioning
The stem bark of Lonchocarpus griffonianus (LGSB) was collected, authenticated, and extracted according to established procedures 8. A portion of the most active extract (130 g) was dissolved in distilled water and exhaustively partitioned with dichloromethane (DCM; 300 mL × 7) in a separating funnel. The resulting fractions were concentrated under reduced pressure to yield the DCM fraction (13.8 g; 10.5%) and the aqueous fraction (105 g; 80%). Both fractions were subjected to biological evaluation for comparative activity as previously reported 8.
Vacuum Liquid Chromatography (VLC) of the Bioactive DCM Fraction
Ten grams of the bioactive DCM fraction was adsorbed onto silica gel (60–120 μm), triturated to form a homogeneous mixture, and subjected to vacuum liquid chromatography. Seven eluates were collected, concentrated in vacuo, and monitored by thin-layer chromatography (TLC) on silica gel GF254 plates using a hexane–dichloromethane (1:4) solvent system. Based on TLC profiles, the eluates were pooled into three fractions: A (fractions 1–3; 5.80 g, 35.5%), B (fraction 4; 0.49 g, 15.7%), and C (fractions 5–7; 2.36 g, 39.3%). Fraction A, which exhibited significant activity, was further purified by gravity column chromatography on silica gel (37 × 2 cm) packed with n-hexane. Elution was carried out using a gradient of n-hexane and ethyl acetate, starting from 100% n-hexane and gradually increasing the proportion of ethyl acetate (99:1, 98:2, 97:3, and 96:4, v/v). A total of 114 eluates (20 mL each) were collected and combined into seven pooled fractions based on TLC profiling (D: 7–19; E: 26–34; F: 35–47; G: 48–64; I: 68–71; J: 75–88; K: 90–114). Further purification of fraction G (2.40 g) yielded compound LO1 (1.92 g), while fraction J (160 mg) furnished compound LO2 (33 mg).
Melting Point Determination
The melting points of LO1 and LO2 were determined on a Kofler hot-stage microscope. Finely powdered samples were sealed in thin-walled glass capillaries, and the temperature range from initial melting to complete liquefaction was recorded.
Spectroscopic Analysis and Structure Elucidation
Compounds LO1 and LO2, both isolated as amorphous white powders, were characterized using electron ionization mass spectrometry (JEOL JMS 600H-1) and one- and two-dimensional nuclear magnetic resonance (1D/2D NMR) spectroscopy (Avance Neo 600 MHz, CDCl?)
3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyl tetrazolium bromide (MTT) Assay
The cytotoxic activities of LO1 and LO2 were assessed using the MTT assay against human prostate cancer (PC3) and cervical cancer (HeLa) cell lines. Cells were seeded in 96-well plates at a density of 1 × 10? cells per well and incubated at 37 °C in a 5% CO? atmosphere for 24 h. Test compounds (30 μM), dissolved in sterile DMSO were added in triplicate. Doxorubicin (30 μM) served as the reference drug. Following 48 h of incubation, MTT solution (200 μL, 0.5 mM) was added to each well, and plates were incubated for an additional 3–4 h. The resulting formazan crystals were dissolved in DMSO (100 μL/well), and absorbance was measured at 570 nm using a microplate reader 10.
RESULTS
Spectroscopic analysis of compound LO1
The molecular ion peak appears at m/z 426.6.3, corresponding to a molecular formula of C30H50O. The loss of a water molecule results in a peak at m/z 408.3. Demethylation of the methyl (CH3) group at C8 produces an ion with m/z 393.3. Further cleavage of an alkane (C2H4) leads to a daughter peak at m/z 365.3. The loss of C13H20 due to McLafferty rearrangement creates a stable peak at m/z 189.3. This pattern reflects the key fragmentation features of a lupane-type triterpenoid molecule. 1D NMR analysis of compound LO1 produced the following spectral data used for its characterization and identification. Comparison with reported literature values revealed similar signals to the seven methyl protons listed in the literature 11 (Table 1): 0.74, 0.76, 0.80, 0.92, 0.94, 1.01, and 1.66. A sextet of a single proton at 2.36 corresponds to the 19 β-H proton and is a characteristic signal of lupeol. The olefinic protons at C29 are a key feature of the lupane-type triterpenoid and appeared at chemical shifts 4.55 and 4.67. The proton attached to C3 appears as a doublet at 3.18. The COSY spectrum of LO1 showed cross peaks between the proton attached to C19 at δ 2.36 and a sp3 methylene proton attached to C21 at δ 1.32, as well as another sp3 methine proton signal at C18 at δ 1.65. There is also coupling between the oxygenated methine proton on C30 at δ 3.16 and a sp3 methylene proton attached to C2 at δ 1.50. The melting point of LO1 was determined to be between 214 °C and 215 °C. This further confirms the identity of the isolated compound.
Table 1 1H NMR (400 MHz) chemical shift pattern of LO1 compared with the reported values in the literature
|
Position |
1H NMR reported in literature |
1H NMR experimental |
Nature of proton |
|
1 |
0.90 (m) |
0.88 (m) |
CH2 |
|
2 |
1.52 (m) |
1.48 (m) |
CH2 |
|
3 |
3.2 (dd) |
3.18 (dd) |
CH |
|
5 |
0.67 (d) |
0.67 (d) |
CH |
|
6 |
1.37 (m) |
1.38 (m) |
CH2 |
|
11 |
1.2 (m) |
1.2 (m) |
CH2 |
|
15 |
1.05 (m) |
1.06 (m) |
CH2 |
|
19 |
2.4 (m) |
2.36 (m) |
CH |
|
21 |
1.36 (m) |
1.32 (m) |
CH2 |
|
22 |
1.37 (m) |
1.3 (m) |
CH2 |
|
23 |
0.90 (s) |
0.94 (s) |
CH3 |
|
24 |
0.76 (s) |
0.74 (s) |
CH3 |
|
25 |
0.83 (s) |
0.80 (s) |
CH3 |
|
26 |
1.01 (s) |
1.07 (s) |
CH3 |
|
27 |
0.94 (s) |
0.92 (s) |
CH3 |
|
28 |
0.79 (s) |
0.76 (s) |
CH3 |
|
30 |
1.67 (s) |
1.66 (s) |
CH3 |
Spectroscopic analysis of compound LO2
The mass spectrum of LO2 showed peaks at an m/z ratio of 414.3, which corresponds to the molecular formula C29H50O. A peak at m/z 396 results from the loss of a water molecule [H2O] from the molecular ion. Further dealkylation [CH3] produced the peak at m/z 381—a peak at m/z 273 results from the fragmentation of the side chain [C10H21] at C17-C20. The dehydration (-H2O) of the daughter fragment at m/z 273 produced the peak at m/z 255. Comparison with reported literature values 12 showed similar signals consistent with the presence of double angular methyl protons attached to C18 and C19, appearing at 0.64 and 1.23 ppm, respectively. The branching methyl protons at C21 were observed at 0.99. Likewise, the characteristic methyl protons of the isopropyl group at C26 and C27 appeared at 0.78 as an overlapping 6H doublet. The methine proton, highly deshielded due to the attached electronegative oxygen atom at C3, appeared at 3.57 as a single proton multiplet.
The overlapping triplet signal at 5.20 indicates olefinic protons at C6 (Table 2). The melting point of LO2 was found to be within the range of 133-135ºC. This further confirms the identity of the isolated compound.
Table 2: 1H NMR (400 MHz) chemical shift pattern of LO2 compared with the reported values in the literature
|
Position |
1H NMR reported in the literature |
1H NMR experimental |
Nature of carbon |
|
1 |
1.08 |
1.10 |
CH2 |
|
2 |
1.49 |
1.49 |
CH2 |
|
3 |
3.53 |
3.57 |
CH |
|
4 |
2.24 |
1.98 |
CH2 |
|
6 |
5.35 d |
5.20 |
CH2 |
|
7 |
1.56 |
1.51 |
CH2 |
|
8 |
1.46 |
1.39 |
CH |
|
9 |
0.94 |
0.95 |
CH |
|
11 |
1.45 |
1.50 |
CH2 |
|
12 |
1.15 |
1.12 |
CH2 |
|
14 |
1.03 |
1.98 |
CH |
|
15 |
1.05 |
1.15 |
CH2 |
|
16 |
1.24 |
1.24 |
CH2 |
|
17 |
1.13 |
1.15 |
CH |
|
18 |
0.68 s |
0.64 |
CH3 |
|
19 |
1.10 s |
1.23 |
CH |
|
20 |
1.36 |
1.25 |
CH |
|
21 |
0.94 s |
0.99 |
CH2 |
|
22 |
1.32 |
1.30 |
CH2 |
|
23 |
1.15 |
1,25 |
CH3 |
|
24 |
0.93 |
0.94 |
CH3 |
|
25 |
1.65 |
1.63 |
CH3 |
|
26 |
0.82 d |
0.83t |
CH3 |
|
27 |
0.83 d |
0.87 d |
CH3 |
|
28 |
1.22 |
1.24 |
CH3 |
|
29 |
0.85 t |
0.82 |
CH3 |
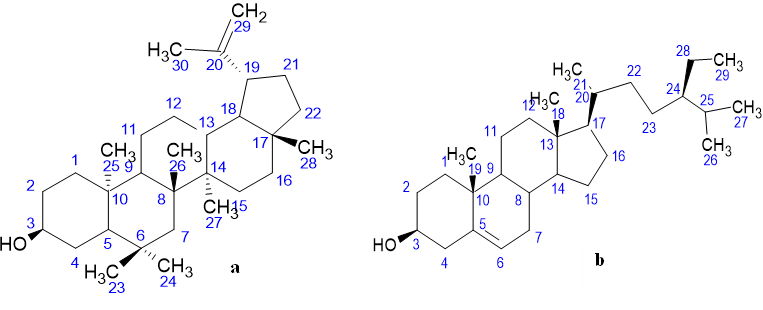
Figure 1: chemical shift allocation for 1HNMR of (a) 3β-lup-20(29)-en-3-ol (Lupeol) and (b) Stigmast-5-en-3β-ol (β-sitosterol)
Table 3: Cytotoxic effect of compounds isolated from LGSB against human cancer cell lines
|
Compound (30 µM) |
Percentage inhibition (%) |
||
|
|
PC3 |
Hela |
BJ |
|
Lupeol |
40.80 |
36.60 |
14.10 |
|
β-sitosterol |
23.60 |
25.10 |
42.70 |
|
Doxorubicin |
89.91 |
90.60 |
89.61 |
PC3 (prostate cancer cells), HeLa (uterine cervical cancer cells), BJ (normal cells)
DISCUSSION
A comparison of the spectral properties of compounds LO1 and LO2 provided a complete elucidation of the compounds. Mass spectrum (MS) fragmentation patterns, along with 1D and 2D NMR chemical shifts, were compared with relevant literature data 13,14. LO1 was identified as lupeol, and LO2 as β-sitosterol. Pentacyclic triterpenoid belongs to a group of compounds now collectively referred to as phytosterols. This class of compounds is unique due to the wide range of pharmacological activities shown by plant species that produce them 15. Structurally, they consist of a system with 30 or fewer carbons, built from isoprene (a 5-carbon unit derived from acetate) building blocks, which originate from squalene. Other compounds derived from squalene include intermediates such as [dammarane, lanostane, oleane (oleanolic acid), lupane (lupeol), ursane (ursolic acid)], or triterpenoid sapogenins like cycloartane, friedelane, filicane, and cucurbitane triterpenoids. These compounds display properties such as anti-cancer, anti-inflammatory, anti-bacterial, antioxidant, anti-viral, anti-lipidemic, and cytotoxic effects 16.
Cytotoxicity measures the level of toxicity of compounds on various cells, utilizing multiple cellular models. Such analyses are typically based on cell cultures with the compound of interest and the measurement of indices associated with cellular growth, such as mitochondrial activity and the ability to undergo cell cycling. Several stains are available to quantify these effects, such as 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) (determination of the oxido-reductive activity of mitochondria), which is the capacity to reduce the stain by living cells. MTT assay has been used to evaluate compounds from the triterpenoid class with significant success 16.
The analysis showed that lupeol suppressed prostate cancer cell growth by 40.8% at 30 µM. This result aligns with the work of Prasad et al. (2018), who demonstrated the effect of lupeol on HeLa cancer cells at a concentration of 100 µM. According to the study, 100 µM caused a 71% inhibition of HeLa cells. A previous report indicated that Lupeol treatment of HeLa cells induced S-phase cell cycle arrest, which may be the mechanism behind the decreased cell growth. The effect of lupeol was observed to reduce the expression of Cyclin E and Cyclin at 24 hours 17.
Several lupeol-expressing plant species have been reported to exhibit anticancer properties, such as lupeol isolated from Chrysanthemum indicum L. (Asteraceae), which displayed a potent cytotoxic effect against human colon adenocarcinoma (SW620) cell lines with an IC50 value of 1.99 μmol/L 18. Pathom Somwong (2017) reported that Ficus racemosa Willd. (Moraceae) with an extract containing high lupeol content exhibited a cytotoxic effect that was directly proportional to the amount of lupeol present in the plant.
β-sitosterol, isolated from L. griffonianus in this research, elicited a 25.1% inhibition at a 30 µM concentration against HeLa cells. This finding is consistent with a 24-hour study that utilized β-sitosterol derived from Pinellas tuber, which demonstrated a 40% reduction in the proliferative activity of HeLa cells at a dose of 20 mM. Another study, reported by Alvarez-Sala et al. (2019), indicated a lower anti-proliferative activity, with 20.5% inhibition at a concentration of 13 mM within 24 hours. 19. Reddy et al. (2022) reported that β-sitosterol has an anticancer effect against prostate cancer cells and can be used to treat cancer with fewer side effects 20. The results suggest that the isolated phytosterols are less toxic to BJ (normal human cell) lines than the standard anticancer drug used as a positive control, further supporting the idea that natural products could be relatively safer. Similarly, Studies have shown that it interferes with multiple cell signaling pathways, including cell cycle regulation, apoptosis, proliferation, survival, invasion, angiogenesis, metastasis, and inflammation. However, most studies are incomplete, partly because β-sitosterol is relatively less potent 21.
ACKNOWLEDGEMENT:
International Centre for Chemical and Biological Sciences (ICCBS), University of Karachi, Pakistan.
REFERENCES

Daniel A. Ambe, Josephine O. Ofeimun, Stephen Okpo, Buniyamin A. Ayinde, Bioactive Sterols from Lonchocarpus griffonianus (Baill.) Dunn: Structural Characterization and Selective Cytotoxic Activity, Int. J. of Pharm. Sci., 2026, Vol 4, Issue 3, 66-73. https://doi.org/10.5281/zenodo.18839772
 10.5281/zenodo.18839772
10.5281/zenodo.18839772
