
We use cookies to ensure our website works properly and to personalise your experience. Cookies policy
Department of Pharmaceutical Chemistry, College of Pharmaceutical Sciences, Government Medical College, Thiruvananthapuram, Kerala, India.
Indole derivatives have emerged as privileged scaffolds in medicinal chemistry due to their structural flexibility and wide-ranging bioactivity. This review highlights recent developments in the synthesis, biological evaluation, and structure–activity relationship (SAR) studies of indole-based compounds with therapeutic potential in treating Alzheimer’s disease, cancer, metabolic and neurodegenerative disorders, infections, and inflammation. Special emphasis is placed on dual/multi-target compounds, advanced synthetic strategies, and molecular docking analyses. Recent studies are discussed in the context of their biological activity, target selectivity, and potential for further optimization.
Indole, a fused heterocyclic structure comprising a six-membered benzene ring and a five-membered nitrogen-containing pyrrole ring, is widely recognized for its prominence in medicinal chemistry. This core framework is found in a variety of natural products and pharmaceuticals due to its adaptability in chemical synthesis and high binding affinity to diverse biological targets [1,2,5]. Modifications at specific positions on the indole scaffold, particularly N1, C3, and the substituted aryl positions, have been shown to substantially alter the pharmacological activity of derived compounds. These structural variations have enabled the development of indole derivatives with a wide range of therapeutic applications, including antioxidant, anticancer, anti-Alzheimer, antiviral, and antimicrobial effects [3,4,6,9]. Recent studies have reported that introducing different pharmacophores such as sulfonamides, pyrazolines, pyridinium salts, and metal complexes to the indole ring can enhance selectivity and potency toward specific biological targets [1,2,6,10]. Some compounds have demonstrated multitarget mechanisms, offering therapeutic benefits in complex diseases. For example, dual inhibitors of cholinesterase enzymes are effective in managing Alzheimer’s disease [1], while simultaneous inhibition of epigenetic regulators such as HDAC and LSD1 presents a promising strategy for cancer therapy [12,13]. Additionally, indole-based antivirals have been designed to disrupt key viral replication enzymes like PA and NP [11]. The integration of in silico methods, including structure–activity relationship (SAR) studies and molecular docking, has played a crucial role in predicting and validating biological interactions. These computational techniques have helped identify essential interactions between indole derivatives and proteins such as EGFR, GyrB, and tyrosinase, supporting their therapeutic relevance [3,5,7]. This review compiles and discusses recent findings on the synthesis and pharmacological applications of indole derivatives, emphasizing their structure–activity relationships and mechanisms of action. The following sections categorize these findings based on disease areas such as neurodegeneration, cancer, microbial infections, oxidative stress, and skin pigmentation disorders, showcasing the broad therapeutic landscape of indole-based drug candidates.
Synthesis And Biological Evaluation of Indole Derivatives
Indole-based compounds have been widely investigated for their therapeutic potential due to their structural versatility and biological activity across a range of targets. This section summarizes recent synthetic strategies and associated biological evaluations of indole derivatives categorized by their pharmacological profiles.
2.1. Anti-Alzheimer’s Agents
Homoud et al. synthesized sulfonamide-substituted indole derivatives by reacting amine-functionalized indoles with various aryl sulfonyl chlorides under basic conditions [1]. Among the synthesized series, compound 9 demonstrated the most potent dual inhibition of acetylcholinesterase (AChE) and butyrylcholinesterase (BuChE), with IC?? values of 0.15 µM and 0.20 µM, respectively. Molecular docking supported these results, showing interactions at both catalytic and peripheral sites of AChE. Farrokhi et al. developed a distinct series of 1-alkyl-4-[(5,6-dimethoxy-1-indanone-2-yl)methylene]pyridinium halide derivatives via aldol condensation followed by N-alkylation. These compounds showed promising dual cholinesterase inhibition, with long-chain alkyl substituents improving enzyme engagement and molecular interaction stability [16].
2.2. Antidiabetic Agents
Zhao et al. developed a series of N-arylsulfonyl-indole-2-carboxamide derivatives via amide bond formation reactions [2]. These compounds were evaluated as fructose-1,6-bisphosphatase (FBPase) inhibitors, a promising target for type 2 diabetes. The most active derivatives displayed strong binding affinities in docking studies with key active site residues (Arg243, Gly259) and were supported by molecular dynamics simulations. Most recently, Ba?c? et al. synthesized a novel series of 5-halogenated N-indolylsulfonyl-2-fluorophenol derivatives through sulfonamide formation. Among them, compounds 3b and 3d exhibited significant aldose reductase inhibitory activity, with IC?? values of 3.14 µM and 1.94 µM, respectively, outperforming the standard inhibitor epalrestat [20]. The presence of electron-withdrawing halogens at the 5-position on the indole ring was associated with improved enzyme binding. Docking analyses confirmed stable interactions with the NADPH binding site of aldose reductase.
2.3. Antioxidant And Cytoprotective Compounds
Aziz et al. synthesized indole-based heterocycles via chalcone intermediates and subsequent cyclization reactions [3]. Compound 10 showed excellent antioxidant potential in ABTS radical scavenging assays, with activity surpassing that of ascorbic acid. Babijczuk et al. reported C3-methylene-bridged indole derivatives synthesized from gramine. These compounds demonstrated cytoprotective activity against oxidative hemolysis in red blood cells and displayed additional antimicrobial activity [4]. Bai et al. designed and evaluated a panel of 3-indolyl-3-hydroxy oxindole derivatives, which showed potent antifungal activity along with moderate antioxidant potential. The substitution pattern at the oxindole moiety was found critical for dual functionality [19].
2.4. CNS Modulators
Ku?aga et al. designed indole–triazine hybrids through formylation of indoles followed by triazine ring construction [5]. These compounds showed nanomolar affinity toward 5-HT? receptors, making them candidates for neuropsychiatric disorders.
Wróbel et al. introduced azaindole–pyrrolidine hybrids that demonstrated multitarget activity by modulating serotonin, dopamine, and norepinephrine receptors, supported by radioligand binding assays and docking studies [6].
2.5. Anticancer Agents
Khalilullah et al. synthesized pyrazolinyl-indole derivatives from chalcones via hydrazine-mediated cyclization [7]. Several compounds, especially HD05, exhibited cytotoxicity across NCI cancer cell lines and targeted EGFR tyrosine kinase.
Al-Wabli et al. reported benzyl-indole carbohydrazides inducing apoptosis via mitochondrial membrane disruption [8]. Aputen et al. developed platinum(IV)–indole prodrugs, which showed nanomolar cytotoxicity and were activated by tumor-specific reduction mechanisms [9]. Zheng et al. synthesized indole–pyridine chalcone hybrids targeting tubulin polymerization and hexokinase 2 (HK2). Compound 26 disrupted cytoskeletal dynamics and energy metabolism, resulting in potent anticancer activity in triple-negative breast cancer cells [17]. Saleh et al. developed 6-indolylpyridone-3-carbonitrile derivatives that inhibited multiple kinases in the MAPK signaling cascade, including EGFR, B-RAF, and CDK4/6, with nanomolar potency. Molecular docking confirmed strong affinity within ATP-binding pockets [18].
2.6. Antiviral Agents
Zhang et al. synthesized benzamide–indole hybrids with dual inhibition of PA and NP subunits in the influenza polymerase complex [12]. Compounds 8e and 8f demonstrated potent antiviral activity, supported by docking and viral replication assays.
2.7. Tyrosinase Inhibitors
Xu et al. designed thiourea–indole derivatives via Schiff base condensation [13]. Compound 4b showed tyrosinase inhibition superior to kojic acid, and docking confirmed effective metal coordination with active site copper ions.
2.8. Antimicrobial And Antibacterial Agents
Babijczuk et al. also evaluated the antimicrobial activity of indoles bearing azole substituents at the N1 position, which exhibited efficacy against both bacterial and fungal pathogens [4]. Li et al. developed indole–pyridinium hybrids by attaching cationic moieties through hydrazide and acrylate linkers [14]. Compound 43 showed exceptional activity against plant pathogenic strains of Xanthomonas oryzae, with in vivo protection and therapeutic effects exceeding 40%. Kong et al. designed pyrimido[4,5-b]indole analogs to inhibit bacterial DNA gyrase and topoisomerase IV (GyrB and ParE) [15]. These compounds were effective against multidrug-resistant Gram-negative bacteria, with favorable in vivo profiles and minimal toxicity. Recent advancements in the synthesis of indole derivatives have led to the development of structurally diverse hybrids exhibiting potent biological activities across multiple therapeutic domains. Various synthetic strategies, such as sulfonamide coupling, chalcone cyclization, and N-alkylation, have yielded compounds with promising anti-Alzheimer’s, antidiabetic, antioxidant, and neuropharmacological properties. Notably, sulfonamide-substituted indoles and pyridinium hybrids showed strong dual inhibition of cholinesterase enzymes, while N-arylsulfonyl and halogenated indolyl derivatives emerged as effective inhibitors of fructose-1,6-bisphosphatase and aldose reductase, respectively. Several antioxidant candidates, including methylene-bridged and thiophene-based indoles, demonstrated cytoprotective and radical scavenging activity. Indole-based scaffolds have also shown significant anticancer, antiviral, and antimicrobial effects. Pyrazoline-bearing indoles and platinum(IV)-indole conjugates exhibited potent cytotoxicity via EGFR inhibition and redox activation. Additionally, novel derivatives targeting tubulin, HK2, and kinases in the MAPK pathway further expanded their anticancer potential. Antiviral benzamide–indole hybrids effectively inhibited influenza replication, while azole-substituted and pyridinium-linked indoles displayed strong antibacterial and antifungal efficacy, including activity against drug-resistant pathogens. These findings underscore the therapeutic versatility of indole scaffolds and highlight their value in future drug design and development.
Table 1. Representative Indole Derivatives Exhibiting Highest Potency Across Therapeutic Categories
|
Compound |
Target / Activity |
IC?? / EC?? |
Therapeutic Area |
Reference |
|
9 |
AChE / BuChE inhibition |
0.15 µM / 0.20 µM |
Alzheimer’s disease |
[1] |
|
3d |
Aldose reductase inhibitor |
1.94 µM |
Type 2 Diabetes |
[20] |
|
10 |
ABTS radical scavenging |
28.23 µg/mL |
Antioxidant |
[3] |
|
HD05 |
EGFR-TK cytotoxicity (leukemia cells) |
Superior to Imatinib |
Anticancer |
[7] |
|
3h |
EGFR, c-MET, B-RAF, CDK4/6 inhibition |
Nanomolar range (multi-kinase) |
Anticancer (MAPK pathway) |
[18] |
|
26 |
Tubulin / HK2 dual inhibition |
Submicromolar against MDA-MB-231 |
Anticancer (dual-target) |
[17] |
|
8f |
Influenza PA and NP inhibition |
Not quantified, potent antiviral |
Antiviral |
[12] |
|
4b |
Tyrosinase inhibition |
Superior to kojic acid |
Skin depigmentation disorders |
[13] |
|
43 |
Antibacterial (X. oryzae) |
>40% in vivo protection |
Antimicrobial (plant pathogen) |
[14] |
|
3u |
Antifungal (R. solani) |
EC?? = 3.44 mg/L |
Antifungal |
[19] |
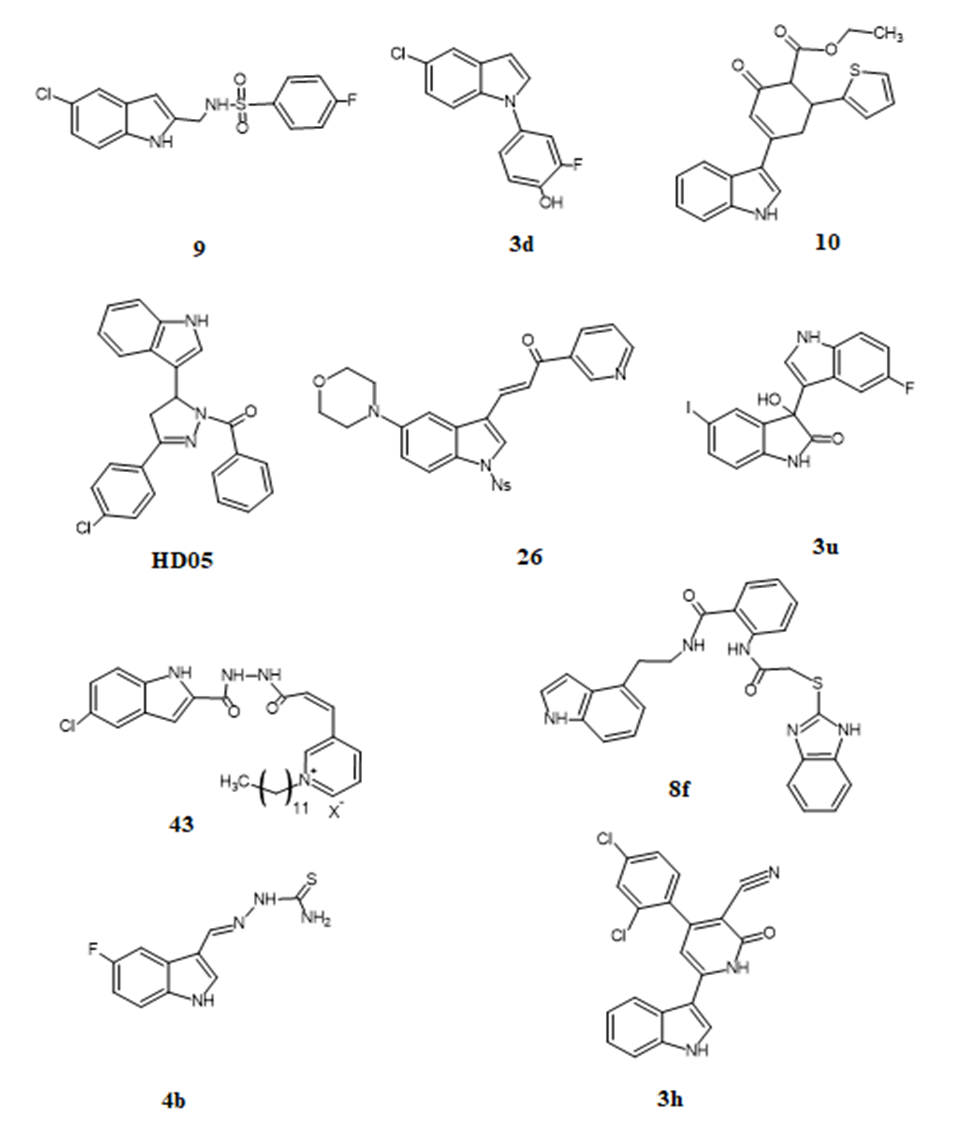
Figure 1. Chemical Structures of Representative Indole Derivatives with Highest Reported Biological Activity.
3. Structure–Activity Relationship (Sar) And Molecular Docking Insights
Structure–activity relationship (SAR) analysis and molecular docking studies offer crucial insights into how specific chemical modifications influence the biological profile of indole-based compounds. Through rational design and computational simulations, researchers have identified key functional groups and structural elements that enhance potency, selectivity, and target interaction across various disease models. A detailed understanding of how chemical modifications influence the biological activity of indole-based compounds has significantly aided the design of more selective and potent drug candidates. Recent studies have illustrated that the incorporation of electron-withdrawing groups, particularly halogens, on the aryl sulfonyl rings of indole sulfonamide derivatives improves inhibition of cholinesterases. For instance, para-chloro substitutions were linked to stronger dual AChE and BuChE inhibition, as confirmed by docking studies that showed favorable π–π and hydrogen bonding interactions within the enzyme's catalytic and peripheral binding regions [1]. In the context of antidiabetic activity, compounds bearing halogenated or bulky aromatic groups on N-aryl sulfonyl indole-2-carboxamides demonstrated enhanced interaction with fructose-1,6-bisphosphatase (FBPase). These structural elements allowed for stable binding to residues such as Arg243 and Gly259, an effect further supported by molecular dynamics simulations [2]. Additionally, 5-halogenated indolylsulfonyl-2-fluorophenols showed excellent inhibition of aldose reductase, a key enzyme implicated in diabetic complications. Docking analyses showed that these compounds formed hydrogen bonds and hydrophobic interactions with critical residues in the NADPH binding domain, with 5-chloro and 5-bromo substitutions offering optimal binding affinity [20]. Antioxidant potential was also influenced by structural variations. Indole derivatives featuring methoxy or methyl substituents on the thiophene ring exhibited improved radical scavenging activity, likely due to increased electron-donating capacity [3]. Similarly, indoles incorporating azole rings at the N1 position were shown to protect red blood cells from oxidative damage, with modeling studies attributing this to high electron density and enhanced redox behavior [4]. In neuropharmacology, halogen substitution on the indole core (particularly at C5) was associated with increased affinity for serotonin receptors such as 5-HT?. This was evidenced by nanomolar binding data and supported by docking simulations [5]. Other indole hybrids containing pyrrolidine or triazine moieties also displayed high affinity across various CNS targets, including dopamine and norepinephrine receptors, where flexibility and lipophilicity appeared to support multitarget binding [6]. With regard to anticancer activity, indole derivatives bearing pyrazoline or carbohydrazide motifs showed promising cytotoxicity profiles. The presence of halogenated phenyl groups and electron-rich side chains improved interactions within the EGFR tyrosine kinase domain and promoted apoptosis via mitochondrial disruption [7,8]. Metal-based conjugates, particularly indole-platinum(IV) hybrids, achieved high potency through tumor-selective activation and controlled release mechanisms. The axial benzyloxy groups on the indole scaffold contributed to improved cellular uptake and activation under reducing conditions [9]. Additionally, recent studies demonstrated that the hybridization of indole with pyridine-based chalcones greatly enhanced tubulin and HK2 inhibition. SAR analysis revealed that electron-donating groups on the phenyl ring improved cytotoxic potency, while docking simulations showed binding at the colchicine-binding site of tubulin and the catalytic pocket of HK2 [17]. Similarly, indole–pyridone derivatives substituted at C6 displayed potent MAPK pathway inhibition, attributed to key interactions with EGFR, CDK4/6, and B-RAF [18]. Dual inhibitors targeting epigenetic regulators such as HDAC and LSD1 have also benefited from SAR optimization. Compounds containing hydroxamic acid or amide functionalities exhibited improved enzyme binding through chelation and hydrogen bonding. In antiviral drug design, benzamide-substituted indoles showed dual engagement with PA-C and NP subunits of the influenza polymerase complex. Electron-withdrawing groups on the phenyl ring were instrumental in enhancing binding affinity and improving antiviral efficacy in vitro [12]. Lastly, in the area of enzyme inhibition, indole–thiourea hybrids exhibited strong activity against tyrosinase. Halogen substituents enhanced the ability of these molecules to coordinate with the copper-containing active site, with docking confirming favorable metal-binding geometries that contributed to improved potency over standard inhibitors [13].
Table 2. Summary of Recent Indole Derivatives: Therapeutic Applications, Key Structural Modifications, and SAR Insights
|
Therapeutic Area |
Modifications |
SAR Insights |
Reference |
|
Alzheimer’s Disease |
Sulfonamide-linked indoles (para-halogenated aryl groups); indanone–pyridinium salts |
Dual AChE/BuChE inhibition; π–π stacking & H-bonds with catalytic/peripheral sites; alkyl chain length ↑ potency |
[1], [16] |
|
Type 2 Diabetes |
N-Arylsulfonyl-indole-2-carboxamides with bulky halogenated rings; 5-halogenated indolylsulfonyl-2-fluorophenols |
FBPase and aldose reductase inhibition; key binding at Arg243/Gly259 and NADPH site; halogens enhance activity |
[2], [20] |
|
Antioxidant Activity |
Indole-thiophene heterocycles; N1-azole groups; oxindole hybrids |
Methoxy/methyl groups ↑ radical scavenging; electron-rich rings protect from hemolysis |
[3], [4], [19] |
|
Neuropharmacology (CNS) |
C5-halogenation; triazine/pyrrolidine hybrids |
High 5-HT7, SERT, D2 affinity; flexible linkers enable multi-receptor binding |
[5,6] |
|
Anticancer (EGFR, HDAC, HK2) |
Pyrazoline ring at C3; carbohydrazides; indole–pyridine chalcones; indolyl-pyridones |
Lipophilic & EWG groups ↑ cytotoxicity; dual kinase inhibition (EGFR/CDK4); ATP-pocket binding |
[7–11], [17], [18] |
|
Antiviral (Influenza) |
Benzamide–indole hybrids with para-electron-withdrawing groups |
Dual inhibition of PA and NP subunits; docking confirms stable binding |
[12] |
|
Tyrosinase Inhibitors |
Thiourea–indole hybrids; halogenated aromatics |
Copper ion chelation; halogenation enhances inhibition over kojic acid |
[13] |
|
Antibacterial / Antimicrobial |
Azole or pyridinium-linked indoles; pyrimidoindoles |
Improved membrane penetration & enzyme binding (GyrB/ParE); in vivo efficacy in rice pathogens |
[14], [15] |
CONCLUSION
Indole derivatives remain at the forefront of medicinal chemistry due to their remarkable structural diversity, synthetic adaptability, and broad spectrum of biological activity. As evidenced by recent studies, modifications to the indole core (such as sulfonamide, pyridinium, pyrazoline, or platinum conjugation) have led to compounds with promising activity across multiple therapeutic domains, including Alzheimer’s disease, cancer, microbial infections, oxidative stress, and skin disorders. Structure–activity relationship (SAR) studies and molecular docking analyses have been pivotal in rationalizing these activities, revealing key interactions like hydrogen bonding, π–π stacking, and metal coordination that govern target selectivity and potency. Moreover, the emergence of multitarget compounds, such as dual cholinesterase inhibitors and epigenetic modulators, underscores the potential of indole-based hybrids in managing complex pathologies. While numerous indole derivatives exhibit strong in vitro and in silico results, their clinical utility will depend on further validation through in vivo studies, pharmacokinetic profiling, and formulation development. Overall, the continued convergence of synthetic innovation, computational modeling, and biological evaluation will ensure the enduring significance of indole scaffolds in the discovery and development of next-generation therapeutics.
Future Perspectives
Despite the significant advancements in the development of indole-based compounds, several critical challenges and opportunities remain for translating these findings into clinical success. As researchers continue to explore this privileged scaffold, the future of indole derivatives in medicinal chemistry appears both dynamic and promising. First, a greater emphasis is needed on multitarget and hybrid compounds, as demonstrated by recent success with dual inhibitors such as HDAC–LSD1 and AChE–BuChE ligands [10,11,16]. These strategies offer an efficient approach to treating multifactorial diseases such as cancer and neurodegenerative disorders. Second, the integration of in silico tools including QSAR modeling, pharmacophore mapping, and molecular dynamics should be paired with experimental validation. Such hybrid computational-experimental workflows can accelerate drug design, as seen in tubulin–HK2 dual inhibitors and FBPase modulators [2,17]. Another vital area is the improvement of pharmacokinetic and pharmacodynamic (PK/PD) profiles, particularly for molecules with high in vitro potency but poor in vivo activity. Innovations such as prodrug approaches (e.g., platinum–indole conjugates [9]) and targeted drug delivery systems may enhance selectivity and minimize off-target effects. Finally, the future development of indole derivatives can benefit substantially from machine learning and AI-driven drug design, enabling rapid prediction of binding affinities, ADMET properties, and synthetic feasibility. Combined with automated synthesis platforms, this will likely reduce the gap between lead identification and clinical translation..
REFERENCES


Akhila S.*, Seema Nair, Exploring the Therapeutic Versatility of Indole Derivatives: Advances in Medicinal Chemistry, Int. J. of Pharm. Sci., 2025, Vol 3, Issue 7, 1543-1552. https://doi.org/10.5281/zenodo.15863064
 10.5281/zenodo.15863064
10.5281/zenodo.15863064