Students, Department of Pharmaceutical Chemistry, Al Shifa College of Pharmacy, Kizhattur, Perinthalmanna
This comprehensive review explores the landscape of Nonsteroidal Anti-Inflammatory Drugs (NSAIDs) and their prodrug variants, elucidating their mechanisms of action, therapeutic applications, and future prospects. Beginning with an overview of traditional NSAIDs and their subclasses, the discussion transitions to various prodrug formulations designed to address limitations such as gastrointestinal irritation and systemic side effects. Examples including NO-NSAIDs, anticholinergic NSAIDs, phospho-NSAIDs, TEMPO-NSAIDs, and HS-NSAIDs are examined, highlighting their unique mechanisms and therapeutic potentials. The abstract concludes by underscoring the transformative promise of NSAID prodrugs in enhancing safety, patient adherence, and personalized medicine, offering insights into the future direction of pain management and inflammation therapy.
NSAIDs, as Nonsteroidal Anti-Inflammatory Drugs, constitute a pharmacological class utilized in alleviating pain, reducing fever, and mitigating various inflammatory conditions. Nonsteroidal anti-inflammatory drugs (NSAIDs) constitute a class of medications endorsed by the FDA for their antipyretic, anti-inflammatory, and analgesic properties [1]. These attributes render NSAIDs valuable in managing a spectrum of conditions including muscle pain, dysmenorrhea, arthritis, fever, gout, migraines, and as adjuncts to opioids in specific acute trauma scenarios [2][3][4]. Stratified according to their molecular composition and specificity, NSAIDs exhibit a diverse array of subclasses, encompassing acetylated salicylates (exemplified by aspirin), non-acetylated salicylates (such as diflunisal and removesalsalate), propionic acids (including naproxen and ibuprofen), acetic acids (such as diclofenac and indomethacin), enolic acids (exemplified by meloxicam and piroxicam), anthranilic acids (comprising meclofenamate and mefenamic acid), naphthylalanine (such as nabumetone), and selective COX-2 inhibitors (like celecoxib and etoricoxib).Moreover, topical formulations of NSAIDs, like diclofenac gel, proffer efficacious relief in acute conditions such as tenosynovitis, ankle sprains, and soft tissue injuries [5][6][7][8]. Herein is cataloged an alphabetical index of FDA-sanctioned NSAIDs:
Table 1: List of NSAIDs
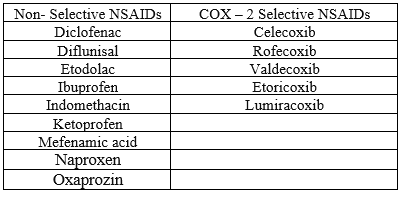
(Note: Rofecoxib and valdecoxib were withdrawn from the market in 2004 and 2005, respectively) [9].
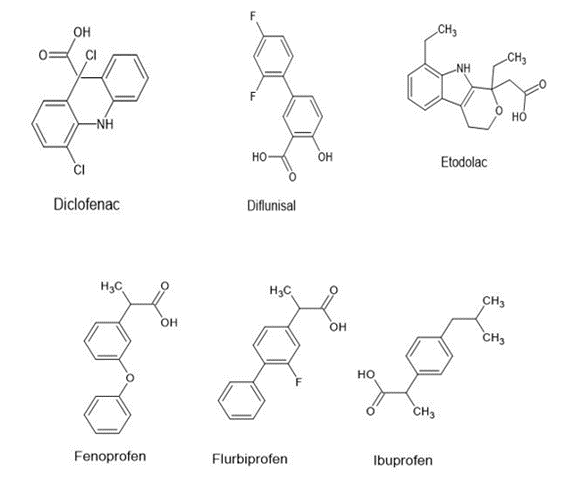
Figure 1.Chemical Structures of some commercially available NSAIDS
The term "prodrug" was introduced into the scientific lexicon by Adrien Albert in 1958, gradually gaining prominence during the 1960s [10,11]. Prodrugs are delineated as altered iterations of drug molecules that undergo biotransformations, either enzymatically or chemically, within the physiological milieu, culminating in the liberation of the active parent compound and thereby precipitating the desired pharmacodynamic response [12]. Fundamentally, while the anticipated pharmacological effect mirrors that of the parent compound, the prodrug itself typically evinces nominal or negligible biological activity. Conventionally, prodrug synthesis entails the covalent conjugation of the parent compound, facilitated with or sans a carrier, to an inert promoiety. Upon administration, this linkage undergoes enzymatic or chemical hydrolysis, liberating the active parent entity [13,14]. In instances where the promoiety possesses pharmacological activity rather than inertness, signifying another drug molecule, a codrug (or mutual prodrug) is engendered [15]. In such instances, pharmacodynamic activity emerges post-enzymatic or chemical cleavage, engendering the emergence of the two parent compounds. NSAID prodrugs of non-selective COX inhibitors were engineered with the primary objective of concealing the unbound acidic moieties within these compounds, thus safeguarding the gastrointestinal tract (GIT) against local irritation. Conversely, the conversion of COX-2 inhibitors into prodrugs was predominantly pursued to yield derivatives boasting heightened aqueous solubility for parenteral administration or to enhance oral bioavailability [16][17][18]. Despite concerted endeavors in the realms of design, synthesis, and evaluation of putatively beneficial NSAID prodrugs, only a scant few exemplars (as depicted in Figure 2) have successfully transitioned into clinical practice, and a subset of these advancements might not have stemmed from a methodical approach to drug development.[19]
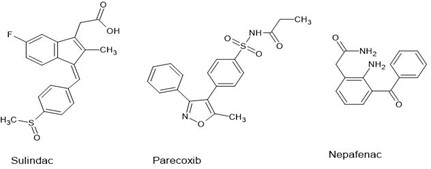
Figure 2 . Chemical Structures of some commercially available prodrugs of NSAIDS
MECHANISM OF NSAID
Prior to 1971, the mechanistic understanding of nonsteroidal anti-inflammatory drugs (NSAIDs) was rudimentary. It was recognized that NSAIDs engendered an anti-inflammatory response that was qualitatively and quantitatively disparate from the more efficacious glucocorticosteroids. Despite extensive documentation of NSAIDs' biochemical effects, hypotheses predicated on these effects had largely been discredited. Among the more tenable postulations of that era posited that NSAIDs, notably salicylates, might attenuate inflammation by inhibiting various proteases. This postulate was corroborated by observations of heightened extracellular proteolytic activity in multiple inflammation models, which was hypothesized to underlie the tissue degradation characteristic of chronic inflammatory pathologies such as rheumatoid arthritis [20-24]
COX-1 AND COX-2
In 1976, a homogeneous, enzymatically active cyclo-oxygenase (COX) or prostaglandin endoperoxide synthase was successfully isolated. This membrane-bound hemo- and glycoprotein, with a molecular mass of 71 kiloDaltons (kDa), was predominantly localized in the endoplasmic reticulum of prostanoid-synthesizing cells. The glycoprotein exhibited COX activity, enabling the cyclization of arachidonic acid and the incorporation of the 15-hydroperoxy group to synthesize prostaglandin G2. The hydroperoxy group of prostaglandin G2 is subsequently reduced to the hydroxy group of prostaglandin H2 by a peroxidase, which employs a diverse array of compounds to furnish the requisite electron pair. Both COX and hydroperoxidase functionalities were integrated within the same dimeric protein molecule [25][26]. This bifunctional enzyme comprises three discrete folding units: an epidermal growth factor-like domain, a membrane-binding motif, and an enzymatic domain. The loci for peroxidase and COX activities are adjacent yet spatially distinct. The elucidation of a membrane-binding motif intimates that the enzyme integrates into a single leaflet of the lipid bilayer, rendering it a monotopic membrane protein. Three helices in the structure constitute the ingress to the COX channel, positing that their insertion into the membrane permits arachidonic acid to access the active site from the bilayer's interior [27]. The X-ray crystallographic structure of COX-2 closely parallels that of COX-1, with analogous binding sites for arachidonic acid on both enzymes. Selectivity for inhibitors is conferred through alternative conformations at the NSAID-binding site within the COX channel, exemplified by a broader entrance and an auxiliary internal pocket [28]. Injury precipitates a multifaceted response in living tissues, termed inflammation, which encompasses the activation of enzymes, the release of signaling molecules, vascular fluid leakage, cellular migration, and the processes of tissue degradation and repair. The cascade initiates with the activation of phospholipase A2, liberating arachidonic acid, which serves as a substrate for cyclooxygenase (COX) or prostaglandin H2 (PGH2) synthase, culminating in an elevated synthesis of prostaglandins. COX is a multifunctional enzyme, endowed with both cyclooxygenase and peroxidase activities. Among the prostaglandins, prostaglandin E2 (PGE2) is the predominant eicosanoid observed in various inflammatory states, ranging from acute edema to chronic arthritis. The prominence of PGE2 as a major product of COX in inflammation suggests that the inflammatory milieu may preferentially direct the enzymatic pathway towards its generation. Additionally, the enzyme 5-lipoxygenase catalyzes the conversion of arachidonic acid into leukotrienes, which play a pivotal role in conditions such as asthma [29].
A substantial array of pharmaceuticals has been developed within this category, constituting a cornerstone therapeutic strategy for the management of acute pain and pyrexia stemming from exacerbated inflammatory responses. An exhaustive understanding of the prostaglandin biosynthetic pathways, the various isoforms of the cyclooxygenase enzyme, and their disparate functions across diverse physiological contexts has significantly expanded the therapeutic applications of these drugs in recent years. Over the past few decades, extensive research has been conducted to elucidate the additional therapeutic benefits of these medications, extending their utility beyond conditions such as arthritis, ductus arteriosus, cancer, and neurodegeneration. Recently, the antimicrobial properties of these drugs have been acknowledged, with potential implications for inhibiting microbial proliferation in the lungs of individuals with cystic fibrosis. Notably, hypertonic ibuprofen formulations have demonstrated significant bactericidal, virucidal, and mucolytic properties, which, when combined with their intrinsic anti-inflammatory effects, may aid in combating infections caused by newly identified enveloped coronaviruses, such as Covid-19 [30]
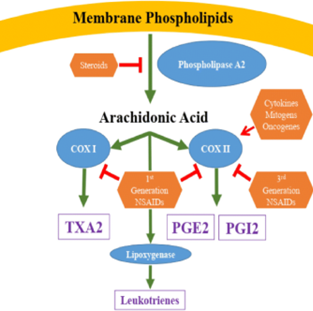
Figure 3 . Mechanism of action of NSAIDS [31].
Assorted Prodrug Variants of NSAIDs
1.NO-NSAIDs
In a synergistic manner, nitric oxide (NO) and prostaglandins (PGs) intricately contribute to the maintenance of gastrointestinal mucosal integrity. This symbiotic relationship suggests that a decrease in the levels of one component prompts a compensatory increase in the other [32]. Consequently, the biosynthetic pathways of these molecules harbor inducible enzymes, notably inducible nitric oxide synthase (iNOS) and COX-2 [33,34]. A multitude of synthetic and pharmacological investigations have explored nitric oxide-releasing prodrugs of NSAIDs (NO-NSAIDs), driven by the rationale that NO may mitigate the diminished protective effects of PGs resulting from NSAID-induced synthesis inhibition. While selective COX-2 inhibitors offer gastrointestinal sparing effects, the withdrawal of rofecoxib and valdecoxib from the market in 2004 due to cardiovascular complications [35] has spurred efforts to discover GI-sparing NSAIDs devoid of selective COX-2 inhibition. Additionally, the appeal of NO-NSAIDs in drug discovery endeavors has surged owing to the well-established vasodilatory properties of NO-releasing agents like nitroglycerin, which can counteract NSAID-induced hypertension [36]. NO-NSAIDs, regarded as codrugs if NO is considered an active pharmaceutical ingredient, have garnered increased attention, often labeled as hybrid prodrugs.[37]
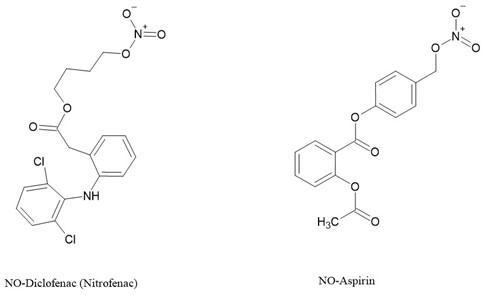
Figure 4. Examples of NO-NSAIDs [37].
2. Anticholinergic NSAIDs and AChEI-NSAIDs
Anticholinergic agents inhibit gastric acid secretion by blocking M1 muscarinic receptors [38] and decrease gastrointestinal motility by targeting M2 and M3 receptors [39]. These effects optimize blood flow and oxygen supply, shielding against ulcers and promoting rapid healing [40][41]. This rationale spurred the development of NSAID prodrugs with localized anticholinergic activity in the gastrointestinal tract prior to absorption [40][42][43]. Conversely, acetylcholinesterase inhibitory NSAID prodrugs are engineered to counteract the inflammatory effects induced by agents like sulfur mustard [44][45]. By modulating the cholinergic anti-inflammatory pathway, these agents enhance acetylcholine availability for receptor binding, exerting potent anti-inflammatory effects [46]. When NSAIDs are transformed into N,N-disubstituted aminoethyl esters, the resultant compounds conform to the classical pharmacophore of anticholinergic agents [19]. Subsequently, these esters release a varying proportion of the parent NSAID in human plasma within a 2-hour timeframe, demonstrating competitive reversible antagonism with pA2 values of ?7.58 compared to 8.20 for atropine sulfate [40][41][42]. The pA2 value signifies the affinity of atropine for its receptor, representing "the negative logarithm of the molar concentration of an equilibrium competitive antagonist that reduces the effect of a double concentration of agonist to that of a single one" [47]. Moreover, these esters exhibit anti-inflammatory efficacy akin to their progenitor NSAIDs while notably mitigating the ulcerogenic index (UI).
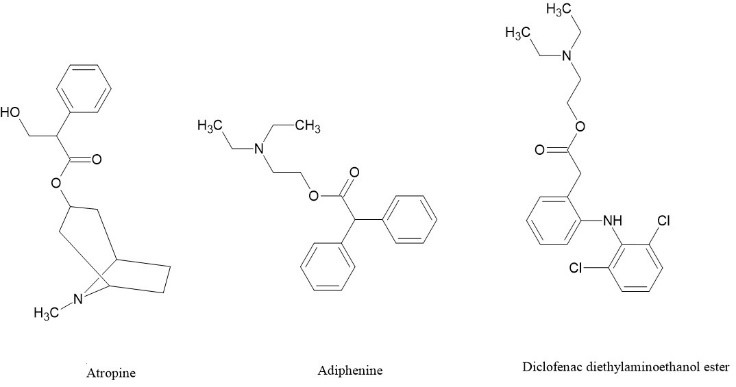
Figure 5 . Examples of Anticholinergic NSAIDS [19].
3. Phospho-NSAIDs
Phospho-NSAIDs represent a fascinating class of compounds comprising an NSAID molecule linked to dialkylphosphate via a linker.Structurally akin to diethylphosphate analogs of nitrate NO-NSAIDs, this group of compounds, particularly phospho-aspirins, has undergone extensive investigation as anticancer agents and, to a lesser extent, as anti-arthritis medications. Although NSAIDs themselves demonstrate antineoplastic properties, their suboptimal efficacy limits their utility as anticancer therapeutics [48]. Phospho-sulindac (PS, OXT-328), phospho-ibuprofen (PI, MDC-917), and phospho-aspirin (PA, MDC-22) have been assessed for their anti-arthritis efficacy, demonstrating effectiveness in alleviating joint inflammation and edema, while exhibiting reduced gastrointestinal toxicity. Notably, OXT-328 and MDC-917 exhibit the ability to suppress PGE2 synthesis in vitro, whereas MDC-22 exclusively inhibits its production in vivo, akin to a genuine prodrug [49]. In the realm of anticancer activity, compounds such as OXT-328, MDC-917 (depicted in Figure 6), have exhibited the capacity to hinder tumor progression through the suppression of cellular proliferation and the facilitation of apoptosis. Remarkably, OXT-328 have demonstrated in vivo efficacy devoid of detectable toxicity in animal models [48]. Phospho-NSAIDs are purported to exert their effects via mechanisms independent of COX, such as the inhibition of the thioredoxin system and the redox-sensitive transcription factor NF-?B [48][50], and/or the induction of reactive oxygen and nitrogen species (RONS)[50]. Phospho-deoxysulindac (OXT-922) impedes the proliferation of human colon cancer cells through a redox/polyamine-dependent mechanism of action [51]. Additionally, MDC-917 demonstrates efficacy against colon cancer [52].
In vivo studies have revealed that carboxylesterases 1 and 2 are capable of hydrolyzing phospho-NSAIDs. Specifically, carboxylesterase-1 exhibits a preference for hydrolyzing phospho-sulindacs, phospho-ibuprofens, phospho-naproxens, and phospho-indomethacins, while carboxylesterase-2 predominantly targets phospho-aspirins. Recent findings indicate that the intact phospho-NSAID molecule is essential for its anticancer activity, and inhibiting carboxylesterases enhances the efficacy of phospho-NSAIDs both in vitro and in vivo [53].
In conclusion, phospho-NSAIDs emerge as promising contenders with potential anticancer and cancer chemopreventive capabilities, serving as significant alternative pharmacological avenues to conventional NSAIDs.
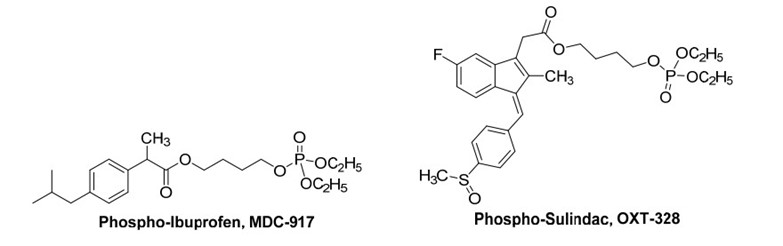
Figure 6 . Examples of Phospho-NSAIDs.
4. TEMPO-NSAIDs
TEMPO (2,2,6,6-tetramethyl-1-piperidinyloxy) and its derivative 4-hydroxy-TEMPO (TEMPOL), as depicted in Figure 7, represent chemically robust nitroxides functioning as nitroxyl radicals, exerting an antioxidative role through radical-radical or radical-scavenging interactions with Reactive Oxygen Species (ROS). In the realm of pharmacology, TEMPOL has exhibited efficacy in mitigating the exacerbation of indomethacin-induced gastric lesions [54]. Flores-Santana et al. (2010) conducted a comprehensive investigation into the synthesis and pharmacological assessment of TEMPOL ester conjugates, namely TEMPOL-ASA (aspirin) and TEMPOL-IND (indomethacin)[55]. The TEMPO-NSAIDs exhibited remarkable abilities to scavenge superoxide radicals, leveraging their TEMPO moieties within the ester structures. Furthermore, both compounds demonstrated potent inhibition of PGE2 production in vitro, with TEMPO-ASA displaying an IC50 of 30 ?g/mL and TEMPO-IND showing an IC50 of 10 ?g/mL, akin to their parent NSAIDs. Notably, their capacity to also inhibit LTB4 production in vitro, a novel pharmacological attribute unique to the intact ester form, underscores their versatility. LTB4, a potent leukocyte activator pivotal in chemotaxis across various inflammatory cascades, underscores the significance of this newfound property, considering the utility of LTB4 receptor antagonists as anti-inflammatory agents [56]. In terms of safety, the maximum tolerable dose (MTD) of TEMPO-ASA mirrored that of aspirin, suggesting a comparable safety profile. Conversely, the MTD of TEMPO-IND exhibited an eight-fold increase, implying a potentially faster in vivo hydrolysis rate than TEMPO-ASA, hinting at the intact molecule's role in the observed safety profile. Further assessment of TEMPO-IND revealed its superiority over indomethacin, boasting a 15% enhancement in in vivo anti-inflammatory activity and approximately tenfold lower ulcerogenic potential [57].
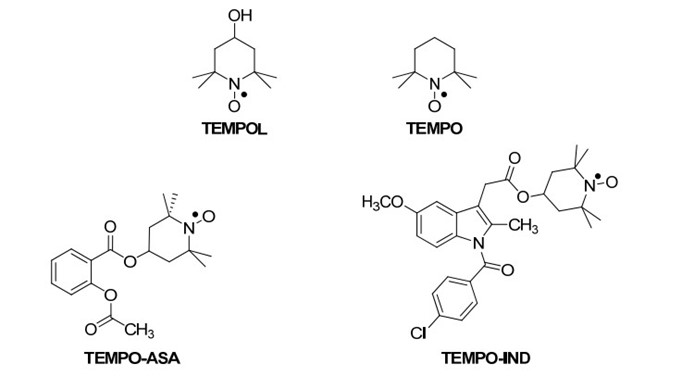
Figure 7. The chemical structures of the TEMPOL esters of aspirin [55].
5.HS-NSAIDs
Physiologically, hydrogen sulfide (H2S) exhibits a dual role in inflammatory responses, with its effects being context-dependent, varying based on the organ and/or dosage administered [58]. Studies have revealed that H2S can mitigate gastric mucosal injury induced by NSAIDs without compromising the drugs' impact on prostaglandin biosynthesis [59]. In fact, H2S donors like 4-hydroxythiobenzamide (4HTB) have been shown to promote more effective ulcer healing [60]. These findings have spurred the development and synthesis of hydrogen sulfide-releasing NSAIDs (HS-NSAIDs). Notably, examples and the mechanism of H2S release [58] from the commonly utilized H2S donating moiety, dithiolethione, are delineated in Figure 24. A captivating instance is exemplified by the salicylic acid ester NBS-1120, meticulously crafted as an H2S- and NO-releasing NSAID, earning the distinguished epithet of a NOSH-NSAID. Noteworthy findings unveil that the potency of H2S donors experiences a remarkable surge when synergistically combined with nitroglycerin, a potent NO donor [60]. While NBS-1120 demonstrates anti-inflammatory efficacy akin to aspirin, its strikingly low IC50 of 48 nM against colon cancer cells signifies an action mediated via a COX-independent mechanism [61]. Conversely, SH-AS manifests considerably diminished activity against the HT-29 colon cancer cell line, boasting a modest IC50 of merely 48 ?M [62]. Subsequent investigations underscore NBS-1120's multifaceted anticancer prowess, elucidating its role in inducing apoptosis and stymieing cell proliferation [63]. Significantly, NBS-1120 exhibits a staggering 1000-fold higher potency in HT-29 cells compared to a composite of aspirin, an H2S donor, and an NO donor, accentuating the indispensability of the intact molecule for its potent anticancer action [63]. Furthermore, NBS-1120 mirrors aspirin's in vivo anti-inflammatory profile, yet surpasses it in inhibiting COX-1, thereby affirming its status as a bona fide salicylic acid prodrug in this domain.
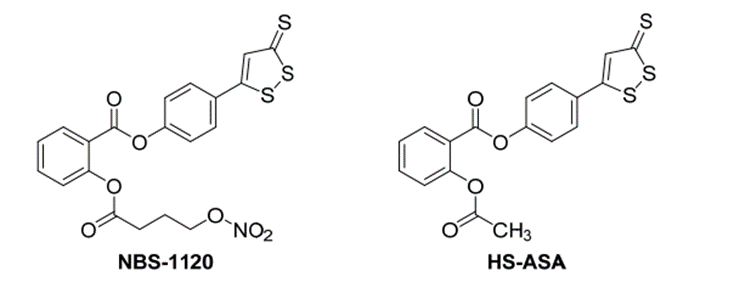
Figure 8.structures of some H2S-releasing NSAIDs (HS-NSAIDs)[19].
FUTURE PERSPECTIVES
NSAID prodrugs hold vast potential across multiple domains, promising advancements in safety, patient compliance, and personalized medicine. Firstly, they offer the tantalizing prospect of mitigating the gastrointestinal complications often associated with conventional NSAIDs. By precisely targeting specific enzymes or receptors, prodrugs can activate at specific sites, thereby reducing systemic exposure and minimizing adverse effects. This targeted approach represents a significant leap forward in drug safety and tolerability. Moreover, prodrugs have the capacity to revolutionize patient adherence to treatment regimens. By extending drug half-life and reducing dosing frequency, they enhance convenience and alleviate the therapeutic
burden, ultimately fostering improved patient compliance and superior clinical outcomes. This aspect is particularly crucial in chronic conditions where long-term adherence to medication is paramount for effective management. In addition, the convergence of pharmacogenomics and advanced drug delivery techniques paves the way for personalized NSAID prodrug therapies. Tailoring prodrug formulations to individual patient characteristics and disease states holds immense promise for optimizing therapeutic outcomes while minimizing risks, ushering in an era of precision medicine in pain management and beyond. Together, these future perspectives underscore the transformative potential of NSAID prodrugs in reshaping the landscape of healthcare.
CONCLUSION
In conclusion, the landscape of NSAID prodrugs presents a multifaceted tapestry of innovation and potential across various therapeutic avenues. These novel formulations offer a glimpse into the future of pharmaceuticals, where safety, efficacy, and patient-centricity converge to redefine the standards of healthcare delivery. The burgeoning field of NSAID prodrugs holds immense promise in addressing longstanding challenges associated with conventional NSAID therapy, particularly concerning gastrointestinal complications and patient adherence. Through precise targeting and controlled drug release, prodrugs aim to minimize systemic exposure and adverse effects while maximizing therapeutic efficacy, heralding a new era of safer and more tolerable pharmacotherapy. Moreover, the advent of personalized NSAID prodrug therapies, facilitated by advancements in pharmacogenomics and drug delivery technologies, represents a paradigm shift towards tailored treatments that optimize outcomes while minimizing risks. By tailoring formulations to individual patient characteristics and disease states, these prodrugs hold the potential to revolutionize pain management and inflammation therapy, ushering in an era of precision medicine in healthcare. As we look to the future, the transformative potential of NSAID prodrugs looms large, promising to reshape the landscape of healthcare delivery and improve the lives of countless patients worldwide. With ongoing research and innovation, the full realization of this potential is within reach, offering hope for a future where safer, more effective, and personalized treatments are the norm.
REFERENCE







Rahila, Shafnaz Abdul Rahman, Rubayyath K. , Digi Davis C. , K. Anuja , Neeshma K. , Nashwa Kattukandathil, Ramsiya K., Razana Binth Yoosuf P., Innovative Horizons In Pain Relief: A Review On The Promise Of NSAID Prodrugs, Int. J. of Pharm. Sci., 2024, Vol 2, Issue 6, 980-993. https://doi.org/10.5281/zenodo.12067690
 10.5281/zenodo.12067690
10.5281/zenodo.12067690
