
We use cookies to ensure our website works properly and to personalise your experience. Cookies policy
1Department of Pharmaceutical Chemistry, Government College of Pharmacy, Bengaluru 560027, Karnataka, India.
2Vishnu Institute of Pharmaceutical Education and Research, Vishnupur, Narsapur 502313, Medak, Telangana, India.
3School of life sciences, JSS Academy of Higher Education and Research (Mauritius) Ltd. Avenue Droopnath Ramphul, Bonne Terre, Vacoas, Mauritius
In our pioneering seminal work on novel cytotoxic potential agents, we are detailing the virtual screening-guided synthesis, characterization, and cytotoxic evaluation of Bis(9-acridinyl) ketone derivatives, specifically Novel N10-substituted 2-fluoro-7-methyl and 2,7-dimethyl Bis-Acridone derivatives. The study substituted Bis-acridone derivatives were investigated as topoisomerase inhibitors. Molecular docking studies against 1TL8 (Topoisomerase I) and 3QX3 (Topoisomerase II?) proteins revealed promising ligand–protein binding interactions among 16 compounds, with four showing high binding affinity for Topoisomerase II? (3QX3). Compounds BFM, BPL, BPR, and BBT exhibited the highest binding affinities, with BFM and BBT displaying notable significant in-vitro cytotoxic effects were observed against Human Breast Adenocarcinoma (MCF-7) cell lines, with IC50 values recorded at 23 ± 1.8 µg and 31 ± 2.2 µg. Furthermore, molecular dynamics simulations validated the stability of these compounds. The in-silico ADME analysis indicated that compound BPL has the potential to penetrate the blood-brain barrier (BBB), while all synthesized compounds were evaluated accordingly demonstrated favorable bioavailability and low toxicity profiles, positioning them as promising candidates for further optimization as topoisomerase inhibitors in cancer therapy.
Cancer remains one of the primary causes of mortality globally, affecting millions annually. The World Health Organization projects that by 2030, there will be approximately 12 million cancer-related deaths worldwide. Breast cancer specifically involves the uncontrolled growth of abnormal cells in the breast, which can lead to tumor formation. If not addressed, these tumors have the potential to metastasize throughout the body, posing a serious threat to life.[1] The genetic mutations that accumulate in cancer cells over time result in altered cellular behavior. This poses significant challenges during treatment, as these mutations can enable cancer cells to develop resistance to therapies, rendering them less effective.[2] The primary goal of chemotherapy is to eliminate cancer cells; however, it is well recognized that these drugs can also harm healthy cells and immune cells, ultimately compromising the body's antitumor response. Significant efforts have been focused on discovering innovative scaffolds for the development of chemotherapeutics aimed at cancer treatment. Despite the availability of numerous drugs, challenges such as drug resistance and limited specificity remain critical obstacles for medicinal chemists.[3][4]
The distinctive planar ring structure of acridone derivatives enables them to function as either topoisomerase poisons or as stabilizers of G-quadruplex DNA structures. Furthermore, their potential inhibitory effects on telomerase and protein kinases may also play a role in their cytotoxic effects in cancer therapy.[5] DNA topoisomerase II, a crucial nuclear enzyme, is responsible for managing DNA topology and chromatin organization within living cells.[6]
The acridone derivative 1,3-bis(9-oxo-acridin-10-yl) propane has been recognized as a potent and poorly reversible modulator of P-glycoprotein (P-gp) mediated transport of vinblastine. [7] [Figure 3] Additionally, fluorinated acridone derivatives have been synthesized and shown to be effective anticancer agents against cell lines such as MCF7, A549, and HT29.[8] A small series of bisacridines, where two acridone units are connected by either a propyl or butyl spacer, were developed and characterized as potential modulators of P-gp-mediated multidrug resistance (MDR). Among these, the propyl derivative (PBA) demonstrated high specificity and efficacy in increasing vinblastine accumulation while inhibiting its efflux in the MDR cell line KB8–5. Notably, the modulation of vinblastine transport by PBA, similar to that of PSC 833, was found to be poorly reversible even after transferring the cells to a medium free of the modulator.[9]
Acridone [figure 2] derivatives are an important class of heterocyclic compounds containing N atom. Acronine was the first natural acridone derivative isolated from the bark of Australian tree Acronychia baurii belonging to the family Rutacea in 1948 and it exhibited potent activity against cancer. In subsequent investigation it was found that several series of analogues of acridone showed potent cytotoxicity. [10] Acridone derivatives disrupt the normal functioning of tumor cells, particularly by affecting enzymes that regulate DNA topology, such as topoisomerases, telomerases, and cyclin-dependent kinases.[11] A study involved synthesizing a substituted acridone with a butyl side chain and acetoxy groups at positions 1 and 3, and its in vitro cytotoxicity was evaluated against the MCF-7 and HL-60 cell lines, revealing moderate cytotoxic effects.[12] Additionally, the introduction of a butyl alkyl side chain at positions 1 and 3 significantly enhanced the cytotoxic activity of the compounds.[13]
Acridone as an anticancer in many cases DNA intercalation,[14][15] Topoisomerase inhibition Topo-II Inhibition,[16][17] Topo-I inhibition,[18][19] Reactive oxygen species inhibition(ROS), [20][21][22] Telomerase inhibitors,[23][24] P13K/AKT/mTOR inhibition, [25][26] PDGFR-? expression,[27] cell cycle arrest, [28] Tubulin Polymerization Inhibition,[29] p53 Activation,[30] CDK Inhibition,[31][32] Histone Deacetylase (HDAC) Inhibition,[33] PARP (Poly ADP Ribose Polymerase)[34] Aromatase Inhibition,[35] etc
In medicinal chemistry, the keto group can contribute to the molecule's ability to form hydrogen bonds and engage in polar interactions with biological targets, enhancing its binding affinity and therapeutic potential. In the case of Bis-acridone compounds often contain a two carbonyl group (keto group) that plays a essential role in their biological activity, such as intercalation with DNA or interaction with enzymes like topoisomerases[36].
The synthesis of 2-methoxy- and 2-methoxy-7-methyl-9(10H)-acridinones was carried out, followed by oxidative coupling to produce the target acridinone dimers.[37] Additionally, bifunctional compounds have been investigated for their potential as antitumor agents, leveraging the ability of acridines and other planar aromatic compounds to interact with DNA and inhibit enzymes. The production methods, pharmaceutical formulations, and applications of substituted 1-amino-4-nitroacridinones as antitumor agents are also discussed. Furthermore, the synthesis of 3,3-bis[7-methoxy-4-nitro-9(10H)-acridinone-1-ylamino]-N-methyl dipropyl amine is detailed.[38][39]
Bis-acridone [Figure 3] a compound consisting of two acridone units linked by a propyl or butyl spacer. Steric hindrance is the main obstacle for Bis-acridone synthesis in the present study, the main aim is to synthesize novel Bis-acridone derivative with ethyl/methyl/butyl chain and characterized by analytical methods, molecular docking studies were performed to evaluate the binding affinity, ADMET and Cytotoxicity studies were evaluated by using MCF-7 cell lines and correlate the parameters to identify the most potent derivative. Steric hindrance is the main obstacle in Bis-acridone synthesis so here we.
MATERIALS AND METHODS:
The chemicals and reagents utilized in the synthesis were of analytical grade and sourced from reputable suppliers. Thin-layer chromatography (TLC) was conducted on pre-coated silica gel plates (Merck GF254), with spot visualization achieved under UV light. The melting points of the synthesized compounds were determined using both Thiele tube apparatus and an AVI digital melting point apparatus. TLC monitoring was carried out on alumina-supported silica gel 60 F254 plates (Merck).
Nuclear magnetic resonance (NMR) spectra, including ^1H (400 MHz) and ^13C NMR, were obtained using a Bruker Spectrospin 400 MHz spectrometer, with dimethyl sulfoxide (DMSO) as the solvent and tetramethylsilane (TMS) as the internal standard. Chemical shifts were expressed in parts per million (ppm, ?) relative to TMS, utilizing CDCl3 and DMSO-d6 as solvents. Coupling constants (J) were reported in hertz, with multiplicities designated as follows: br (broad), s (singlet), d (doublet), t (triplet), and m (multiplet). Mass spectra of the compounds were recorded using a Waters Mass Spectrometer, with DMSO serving as the solvent. Column chromatography was performed on silica gel with a mesh size of 100–200 (Bruker)
EXPERIMENTAL:
IN SILICO DOCKING STUDIES:
The docking studies for the research work were conducted using the glide tool from Schrodinger molecular drug discovery suite (Maestro version 13.0).
Preparation of ligands:
The structures of the selected ligands were generated using ChemDraw Professional software. These structures were imported into the project table of Maestro in the form of Simplified Molecular Input Line Entry (SMILES). Co-crystal ligand and the standard drug Isocarboxazid SMILES were imported from the PubChem database. All imported ligands were generated using the LigPrep interface, employing the OPLS (Optimized Potentials for Liquid Simulations) 2005 force field at a pH of 7.0 ± 0.2. The calculations were performed with Epik, which is designed to provide precise pKa values in both water and DMSO.Stereoisomer computation was left at “retain specific chirality (vary other chiral centers)”. Generation of stereoisomers of at most 16 per ligand was selected.
Protein preparation:
The topoisomerase proteins selected for this study, with PDB IDs 1TL8 and 3QX3, were obtained from the Protein Data Bank and added to the project table. These proteins may contain heavy atoms, water molecules, cofactors, metal ions, and may exist as multimers. To prepare for docking studies, it is essential to refine the raw 3D structure to ensure it is biologically relevant and suitable for analysis. To achieve this the protein has to undergo protein preparation process. The protein preparation was performed using “protein preparation wizard” available in the glide tool through the Maestro interface. Water molecules were eliminated, and charges along with bond orders were assigned. Additionally, hydrogen atoms were added to the heavy atoms in the structure. The proteins were optimized (H- bond optimization) and minimized (energy minimization).
Molecular docking:
The prepared ligands and protein were docked using the Glide tool. The ligands were positioned in the active site of the target protein employing the standard precision (SP) algorithm, allowing for flexible ligand sampling. The interactions were evaluated using the Glide score, which reflects the optimal fit between the ligand and the receptor. Ligands were ranked according to their Glide scores, with the most negative values indicating better fits. The ligand interaction tool was then used to visualize the interaction diagram, highlighting the ligands interactions with the active site residues of the target protein. The docking score was found to be in the range of -9.884 to -3.912.
GENARAL PROCUDURE:
(I) Synthesis of (BPL) 2-(phenylamino) benzoic acid / (BPR) 5-methyl-2-(p-tolylamino) benzoic acid / (BBT) 2-(phenylamino) benzoic acid / (BFM)2-((4-fluorophenyl) amino)-5-methylbenzoic acid
A mixture of 5 g (0.031 mol) of 2-bromo-5-methylbenzoic acid and 4 g (0.031 mol) of 4-fluoroaniline was placed in a round-bottom flask. To this, 0.20 g (0.0017 mol) of copper powder was added along with 30 mL of isoamyl alcohol, and the mixture was thoroughly mixed. Slowly, 6 g (0.041 mol) of potassium carbonate (K2CO3) was introduced into the reaction mixture. The contents were then allowed to reflux for 6 to 8 hours. After refluxing, isoamyl alcohol was removed via distillation, and the resulting mixture was poured into 1 L of hot water and acidified with concentrated hydrochloric acid. The precipitate formed was filtered, washed with hot water, and collected. The crude product was then dissolved in an aqueous sodium hydroxide solution, boiled with activated charcoal, and filtered. Upon acidifying the filtrate with concentrated hydrochloric acid, a precipitate was formed, which was washed with hot water and recrystallized from methanol, yielding a greyish solid. The progress of the reaction was monitored using TLC with a mobile phase of petroleum ether and ethyl acetate in a 1:1 ratio, and the spots were visualized under UV light.
(II) Synthesis of (BPL)acridin-9(10H)-one, (BPR)2,7-dimethylacridin-9(10H)-one, (BBT)acridin-9(10H)-one, (BFM)2-fluoro-7-methyl acridin-9(10H)-one
A total of 5 g (0.0016 mmol) of 2,7-(4-methylanilino) benzoic acid was placed in a flask, to which 20 mL of polyphosphoric acid was added. The mixture was heated in a water bath at 100°C for 4 hours. The appearance of a yellow color indicated the completion of the reaction. Following this, the mixture was poured into 1 L of hot water and made alkaline using ammonia solution, resulting in the formation of a greenish precipitate. This precipitate was then filtered, washed with hot water, and collected. The resulting compound was recrystallized using glacial acetic acid. The progress of the reaction was monitored by thin-layer chromatography (TLC) with a mobile phase consisting of petroleum ether and ethyl acetate in a 1:1 ratio, and the spots were visualized under UV light.
(III) Synthesis of (BPL)Bis-acridin-9(10H)-one, /(BPR)10,10'-(propane-1,3-diyl)bis(2,7-dimethylacridin-9(10H)-one)/,(BFM)10,10'-(propane-1,3-diyl)bis(2-fluoro-7-methylacridin-9(10H)-one)/,(BBT)10,10'-(butane-1,4-diyl)bis(2,7-dimethylacridin-9(10H)-one)
One gram (0.0046 mol) of 2-methyl acridone-9(10H)-one was dissolved in 25 mL of tetrahydrofuran. To this solution, 17 mL of 6N potassium hydroxide and 0.43 g (0.0013 mol) of tetrabutylammonium bromide were added. The reaction mixture was stirred at room temperature for 30 minutes. Subsequently, 1,3-dibromopropane, 1,4-dibromobutane, or 1,5-dibromoethane (0.72 g/0.87 g, 0.0063 mol) was added slowly to the mixture, which was then stirred for an additional 24 hours at room temperature. The progress of the reaction was monitored using thin-layer chromatography (TLC). Upon completion, the mixture was poured into ice-cold water, resulting in the formation of a precipitate. This precipitate was filtered, washed with cold water, and air-dried for further use. The tetrahydrofuran was evaporated, and the aqueous layer was extracted with chloroform. The chloroform layer was washed with water, dried over anhydrous sodium sulfate, and concentrated using a rotary evaporator. The final product was characterized by TLC.
Synthesis of target compounds:
BPL 2-methyl-10-[4-(2-methyl-acridone) butyl]acridin-9(10H)-one
Yield: 43.4%; mp: 293°C, Rf: 0.741, Colour: light grey Solid,
1H NMR: ? 2.34 (4H, t, J = 7.2 Hz) (alkyl chain, 4H)., 7.10 (4H, ddd, J = 8.4, 1.3, 0.5 Hz), 7.27-7.43 (8H, 7.34 (ddd, J = 7.9, 7.4, 1.3 Hz), 7.37 (ddd, J = 7.9, 1.3, 0.5 Hz)), 8.12 (4H, ddd, J = 8.4, 7.4, 1.3 Hz) ( aromatic, 16H).13C NMR(400MHz, CDCI3) ?=45-50 (2C, alkyl C), ?=114.1-145 (24C aromatic C), ?= 178(C=O, keto 2C) MS(+ESI) m/z % (M+H) = 416.12. Found:417.12,
BPR 10,10'-(propane-1,3-diyl)bis(2,7-dimethylacridin-9(10H)-one)
Yield: 49%, M.P: above 300°C, Rf: 0.841, Color: Dark brown Solid,
1H NMR: ? 1.94 (2H, quint, J = 6.7 Hz) (acyl chain, 2H), 2.33 (12H, s) (CH3 attached to aromatic ring, 4.33 (4H, t, J = 6.7 Hz), 7.00-7.18 (8H, 7.06 (dd, J = 8.2, 1.6 Hz), 7.32 (dd, J = 8.2, 0.5 Hz)), 7.62 (4H, dd, J = 1.6, 0.5 Hz) (aromatic, 12H).13C NMR(100MHz, CDCI3) ?= 14.94 (CH3 attached to aromatic ring, 4C), ?= 18-35(3C, alkyl chain), ?=110-141.265(24C aromatic C), ), ?= 178(C=O, keto ,2C),MS(+ESI) m/z % (M+H) = 486.23 Found:487.12,
BBT 10,10'-(Butane-1,3-diyl)bis(2,7-dimethylacridin-9(10H)-one)
Yield: 47%, M.P: above 300°C, Rf: 0.741, Colour: light grey Solid,
1H NMR: ? 1.12 (12H, tt, J = 7.1, 7.0 Hz), 2.45 (5H, s) (alkyl chain), 5.10 (3H, t, J = 7.0 Hz), 7.00-8.0 (8H, 7.16 (dd, J = 8.2, 1.5 Hz), 8.0 (dd, J = 8.2, 0.5 Hz)), 8.20 (4H, dd, J = 1.5, 0.5 Hz) (CH3 attached to aromatic ring).13C NMR(400MHz, CDCI3) ?=24.2,25.98 (2C S, alkyl C), ?= 59.01(2C S, hetero C-N), ?=114.1-141.255(24C S, aromatic C), ?= 21.14(CH3 attached to aromatic ring(4C S), ?= 178(C=O, keto, 2C S),MS(+ESI) m/z % (M+H) =502.45 Found:503.82,
BFM 10,10'-(propane-1,3-diyl)bis(2-fluoro-7-methylacridin-9(10H)-one)
Yield: 42%, M.P : above 300°C, Rf : 0.641, Color : light grey Solid,
1H NMR: ? 1.50 (2H, quint, J = 7.1 Hz), 2.34 (6H, s) (CH3 attached to aromatic ring), 4.30 (4H, t, J = 7.1 Hz) (CH3 attached to aromatic ring), 7.10-7.23 (6H, 7.16 (dd, J = 8.2, 1.4 Hz), 7.18 (dd, J = 8.2, 0.5 Hz) aromatic, 7.20 (dd, J = 8.7, 1.6 Hz)), 7.40 (2H, dd, J = 8.7, 0.4 Hz), 8.32 (2H, dd, J = 1.4, 0.5 Hz), 8.83 (2H, dd, J = 1.6, 0.4 Hz).13C NMR(400MHz, CDCI3) ?=26.54 (1C, alkyl C), ?= 49.16(2C, hetero C-N), ?=78.420(1C ), ?=115.679 -142.565(26C aromatic C), ?= 21.3(CH3 attached to aromatic ring(2C), ?= 178(C=O),MS(+ESI) m/z % (M+H) = 496.25.Found:497.36,
Cytotoxicity assay:
Cytotoxicity evaluations were performed at the Vishnu Institute of Pharmaceutical Education and Research (VIPER) in Telangana. The MTT assay was employed to assess the cytotoxic effects of synthesized bis-acridone derivatives on MCF-7 breast cancer cells. Initially, MCF-7 cells were seeded in 96-well plates at a density of 5,000 to 10,000 cells per well, using Dulbecco's Modified Eagle Medium (DMEM) supplemented with 10?tal bovine serum (FBS) and 1% penicillin-streptomycin. This media formulation provided essential nutrients and antibiotics to promote optimal cell growth while minimizing the risk of bacterial contamination. After cell seeding, the plates were incubated in a humidified environment with 5% CO? at 37°C for 24 hours to allow for cell attachment and growth.
After the cell attachment period, the cells were treated with various concentrations of the bis-acridone derivatives, ranging from 0.1 to 100 ?M. Doxorubicin was used as a positive control to compare the cytotoxic effects of the test compounds. The treatment duration was set for 72 hours to provide adequate time for the compounds to interact with the cells, facilitating an assessment of their potential cytotoxicity.
At the end of the treatment period, 10 ?L of MTT solution (5 mg/mL in phosphate-buffered saline) was added to each well. The plates were then incubated for an additional 4 hours, allowing viable cells to convert the MTT into insoluble formazan crystals, which serve as an indicator of metabolic activity. After incubation, the culture medium was carefully removed, and 100 ?L of dimethyl sulfoxide (DMSO) was added to each well to dissolve the formazan crystals. This resulted in a purple solution, the intensity of which correlates with cell viability. The absorbance of each well was measured at 570 nm using a microplate reader, enabling quantification of the color intensity, which is directly proportional to the number of viable cells.
IC?? values, representing the concentration of each bis-acridone derivative that led to a 50% reduction in cell viability compared to untreated controls, were calculated. These IC?? values, expressed in micromolar (?M), facilitated a comparative analysis of the cytotoxic potency of the bis-acridone derivatives against the MCF-7 cell line, providing insights into their potential as anticancer agents.
Molecular dynamics simulation
Molecular dynamics simulations of the selected ligands in the 3QX3 complex were conducted using Desmond. The ligand molecules were introduced into the Desmond environment, where force field parameters were assigned utilizing the OPLS force field. To assess the stability of the ligands, different solvent models were employed, specifically incorporating water using the TIP4P-D model. The overall charge of the periodic cubic box was neutralized by adding sodium (Na?) and chloride (Cl?) ions, resulting in a final system comprising approximately 60,000 atoms in the TIP4P-D configuration.
Subsequently, the prepared system underwent a series of steps, including energy minimization, followed by NPT (constant number of particles, pressure, and temperature) and NVT (constant number of particles, volume, and temperature) simulations. The relaxed system was simulated for a duration of 100 ns under NPT conditions, maintaining a temperature of 310 K and a pressure of 1 bar. Atomic coordinate data and system energies were recorded every 250 ps. The trajectory analysis for the ligand involved evaluating the root mean square deviation (RMSD), radius of gyration (Rg), and the number of inter-molecular hydrogen bonds (Inter H-Bonds).
Pharmacokinetics and Physicochemical Properties Prediction:
In the pharmaceutical industry, assessing the drug-likeness, pharmacokinetic, and toxicity profiles of lead compounds is essential to enhance bioavailability and minimize toxicity. To evaluate these aspects, we utilized the web-based Swiss-ADME and pkCSM tools to analyze selected bioactive molecules isolated from Mimosa pudica Linn. These platforms provided insights into the compounds' absorption, distribution, metabolism, and excretion (ADME) characteristics, as well as their safety and physicochemical properties.
RESULTS:
Molecular Docking studies
The in-silico studies were carried out for 25 compounds using the glide tool from the software Schrodinger Maestro (version 13.0) The ligands were docked with the RCSB protein data bank topoisomerase proteins TOPO I(PDB ID: ITL8) and TOPO II Beta (PDB ID: 3QX3).
All synthesized compounds underwent molecular docking studies. The compounds exhibited binding energy in the range of -9.884 to -3.423 (Table No 2) with Doxorubicin as Standard. The compounds have shown interactions like Vander Waals Interaction, pi-pi interaction, alkyl interaction, conventional hydrogen bond. Out of 16 compounds we have selected 4 (BPL, BPR, BBT, BFM) (FIGURE 4 And 5) interaction with protein -ligand compounds based on their affinity towards topoisomerase. The compounds BFM and BBT showed strong interactions with Topoisomerase II, exhibiting binding energies of -9.27 and -9.11 respectively.
Chemistry :
In the current research study, a series of novel Bis-acridone derivatives were synthesized via three step chemical reaction.[39] Acridone / 2-fluro-7-methyl acridone/ 2,7-dimethyl acridone with Ethyl, propyl and butyl chain were synthesized according to [Scheme1]. substation position(Table no 1 )
Parent acridone, 2-fluoro-7-methyl acridone, and 2,7-dimethyl acridone were synthesized through the Ullmann condensation reaction involving 2-chlorobenzoic acid and aniline, 2-bromo-5-methyl benzoic acid and 4-fluoroaniline, as well as 2-bromo-5-methyl benzoic acid and toluidine. This process yielded diphenylamine-2-carboxylic acid, 2-fluorodiphenylamine-2-carboxylic acid, and 2,7-dimethyldiphenylamine-2-carboxylic acid.
Subsequently, cyclization was performed using polyphosphoric acid in a water bath at 100°C, although alternative reagents such as POCl3 and H2SO4 could also be employed to produce the target acridone derivatives. The N-alkylation of the synthesized acridones was achieved using a phase transfer catalyst (PTC) like tetrabutylammonium bromide (TABA). This was necessary due to the nitrogen atom in the acridone structure being relatively unreactive toward N-alkylation with alkyl halides, owing to its weakly basic nature. The reaction involved stirring at room temperature with the alkylating agents dibromo-(CH2)3-Br or diiodo-(CH2)2-I in a biphasic system comprising tetrahydrofuran (THF) and a 6N aqueous potassium hydroxide solution, resulting in the successful synthesis of bis(9-acridinyl) ketone, 2-fluoro-7-methyl bis-acridone, and 2,7-dimethyl bis-acridone with favourable yields.
Recrystallization should be done for products in each step and dried in the hot air oven. The synthesized and purified products will be confirmed by 1H-NMR, 13C-NMR and MASS spectroscopic methods.
CYTOTOXICITY:
The anticancer activity of the synthesized substituted bis-acridone derivatives (BPL, BPR, BBT, BFM) was assessed using the MTT assay (3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyl-tetrazolium bromide) on the MCF-7 cell line. Doxorubicin served as the positive control in this study. Cytotoxicity was measured through the determination of the IC50 value, indicating the concentration required to reduce cell viability by 50%. The treatment involved nine different concentrations of the test compounds, ranging from 0.1 to 100 ?M (specifically, 0.1, 0.5, 1, 2.5, 5, 10, 25, and 50 µM) as detailed in (Table 3). The in vitro cytotoxicity results are presented as the concentration of each compound needed to achieve 50% inhibition of cell growth compared to the control (IC50 ?M).Graphical representation of compounds (figure no: 6)
The results demonstrated that compounds BFM and BBT exhibited superior cytotoxicity compared to BPR and BPL. The IC50 values for BFM and BBT were determined to be 22 ± 3.2 µM and 24 ± 5.2 µM, respectively. In contrast, the IC50 values for BPR and BPL were found to be 32 ± 4.2 µM and 38 ± 2.2 µM, respectively. The positive control, doxorubicin, exhibited an IC50 value of 11 ± 2.2 µM.
The variations in cytotoxicity observed among the synthesized compounds can be linked to the presence of electron-withdrawing groups and differences in chain length. It has been demonstrated that the addition of electron-withdrawing groups to the molecular framework can significantly improve the cytotoxic activity of compounds. In this study, compounds BFM and BBT, which possess more potent electron-withdrawing groups, demonstrated significantly higher cytotoxicity compared to BPR and BPL.
Furthermore, the chain length of the compounds also influenced their cytotoxic activity. As the chain length increased, the inhibitory action of the derivative compounds against MCF-7 cells was observed to improve. This suggests that a longer chain length may facilitate better interaction with the cellular targets, leading to enhanced cytotoxicity.
Pharmacokinetics and Physicochemical Properties Prediction:
Evaluation of ADMET Properties:
Evaluating the ADMET (Absorption, Distribution, Metabolism, Excretion, and Toxicity) characteristics of newly synthesized compounds is crucial in the drug development process. These properties play a significant role in influencing the efficacy and safety profiles of potential therapeutic agents. Understanding how a compound behaves in biological systems helps in predicting its clinical performance and potential side effects. For the bis-acridone derivatives synthesized in this study, a comprehensive assessment of ADMET properties was conducted using the Swiss ADME online platform. This analysis is pivotal in predicting how these compounds behave in biological systems and their potential therapeutic applications. The evaluation covered several key toxicological aspects, including the potential for tumorigenicity, mutagenicity, irritation, and reproductive toxicity. Assessing these properties is crucial for ensuring the safety of the compounds and identifying any adverse effects that could arise during their clinical use. The findings indicated that the ADMET characteristics of the synthesized bis-acridone derivatives were well within the acceptable safety margins, suggesting their viability as drug candidates. The detailed results of the ADMET analysis are presented in[ Tables 4, 5, and 6].
QSAR Analysis:
Alongside ADMET evaluations, quantitative structure-activity relationship (QSAR) studies were conducted to gain deeper insights into the pharmacokinetic and pharmacodynamic characteristics of the synthesized compounds. ADMET properties are fundamental determinants of whether a drug candidate can progress to clinical trials, and oral bioavailability is particularly important, as it reflects the relationship between solubility and permeability of the compound. For the QSAR studies, various computational tools were utilized, with Molinspiration being a significant resource for evaluating the drug-likeness of the bis-acridone derivatives. The analysis included applying Lipinski's rule of five, a well-established guideline for assessing the drug-likeness of compounds based on their physicochemical properties. According to this rule, an ideal drug candidate should possess no more than one of the following characteristics: a molecular weight exceeding 500 Daltons, a log P value Higher than 5, greater than 5 hydrogen bond donors, or more than 10 hydrogen bond acceptors.
This framework enables researchers to identify which compounds are most likely to be orally bioavailable and therapeutically effective. Additional parameters computed during the QSAR analysis included the partition coefficient (log P) and predicted percentage absorption, both of which provide insight into how the compounds will behave within the human body. The results of these QSAR studies, showcasing the favourable attributes of the synthesized derivatives, can be found in [Table 7].
Assessing Bioactivity and Toxicity Risks:
Evaluating the bioactivity and potential toxicity of drug candidates is critical for advancing their development. The QSAR descriptors for the bis-acridone derivatives were predicted using the Molinspiration online server, version 2018.10. This platform allows for the assessment of various chemical and biological properties, including bioactivity scores, which are indicative of a compound's likelihood to exhibit desired therapeutic effects. The outcomes suggested that the synthesized compounds possess a good safety profile, enhancing their candidacy for future clinical applications.
To complement the QSAR findings, the Osiris Property Explorer was utilized to investigate potential toxicity risks and other essential drug characteristics. This tool is effective in identifying undesirable properties that could affect the safety and efficacy of the compounds. The analysis revealed that the synthesized bis-acridone derivatives exhibit favourable profiles regarding toxicity, further supporting their potential for medicinal use.[Figure no:7]
BOILED-egg diagram analysis :
The BOILED-egg diagram analysis, illustrated in [Figure no:8], indicates that the compound lies within the acceptable range for standard drugs. A point within the yellow region suggests that the compound is not impacted by CNS P-glycoprotein, while a red dot in the central 'yolk' area signals passive permeation through the blood-brain barrier (BBB), implying retention in the brain. In this study, the synthesized ligand and its complexes meet these criteria, showing favorable bioavailability. The findings suggest that the compound is safe with minimal risk of causing skin allergies, and the drug scores are notably moderate.
Molecular Simulation:
Molecular dynamics (MD) simulations conducted in a solvent environment under constant temperature and pressure conditions have provided insights into the conformational changes of the Topoisomerase-II B (3QX3) in complex with BPL, BFM, and BBT. Complex stability was assessed through the root mean square deviation (RMSD) and root mean square fluctuation (RMSF) of both protein and ligand components. Compared to the loop regions, alpha helices and beta strands exhibited greater rigidity and consequently lower fluctuation levels. Notably, the loop region of the 3QX3 complex with BPL, BBT, and BFM displayed higher fluctuations, measuring 11.5 Å, 12 Å, and 10.5 Å, respectively. Distinct RMSD patterns for both the protein and ligands were observed across the 100 ns simulation period (see Figure 9). For the 3QX3-BPL complex, an initial increase in RMSD was noted in the first 20 ns, with further drift from 40 to 60 ns, eventually stabilizing between 85 and 90 ns. In the 3QX3-BFM complex, stabilization occurred after approximately 18 ns, with RMSD fluctuations in the ligand observed around 10.5 Å up to 100 ns. The 3QX3-BBT complex exhibited fewer stable contacts, with interactions found to be relatively low throughout the simulation.
Analysis of protein-ligand contacts in the 3QX3-BPL, BFM, and BBT complexes during the 100 ns timeframe is shown in [Figure 10]. Key interacting residues included LYS 425 for BPL (30%), and for BFM, LYS 739 (40%), ASN 786 (60%), and GLN 742 (60%). In the 3QX3-BBT complex, HIS 775 (32%) and ALA 779 were the most significant contact residues [Figure11] shows the evolution of protein-ligand interaction contacts over the 100 ns period. Hydrophilic interactions contributed prominently to binding affinity, with water bridges observed for BPL (10 bridges) at ARG 364, GLU 418, GLN 421, SER 423, ILE 424, LYS 425, ASN 491, LYS 498, and GLU 494; for BFM (12 bridges) at residues such as ARG 503, LYS 505, ILE 506, LEU 507, and ASN 786; and for BBT (10 bridges) including ARG 503, GLY 504, LYS 505, and GLU 777. The overall interaction profile for these complexes throughout the simulation is depicted in [Figure 12].
The ligand torsion profiles of 3QX3 in complex with BPL, BFM, and BBT differed markedly. Radial plots in [Figure 13] illustrate torsion dynamics, with the ligand torsion for BPL between the pyridine ring and carbonyl group showing divergence that likely contributed to extended conformational shifts between 1-3 ns. The molecular properties of compound 3e, including ligand RMSD, radius of gyration (rGyr), intramolecular hydrogen bonds, molecular surface area (MolSA), solvent-accessible surface area (SASA), and polar surface area (PSA), were monitored over 100 ns, as depicted in [Figure14]. Overall, compound 3e showed stable properties with no major fluctuations throughout the simulation.:
This study presents the synthesis and assessment of new bis-acridone derivatives, investigated as potential anticancer agents with a specific focus on targeting breast cancer. The alarming rise in cancer incidence and the associated mortality rates underscore the urgent need for innovative therapeutic strategies. Our findings contribute to this endeavour by exploring the unique properties of bis-acridone compounds, which exhibit promising cytotoxicity against MCF-7 breast cancer cells.
The synthesis of Novel bis-acridone derivatives was successfully achieved, overcoming the challenges posed by steric hindrance associated with the ethyl chain. The incorporation of various substituents, particularly electron-withdrawing groups, was pivotal in enhancing the cytotoxicity of the synthesized compounds. The results demonstrated that compounds BFM and BBT exhibited significantly lower IC50 values (22 ± 3.2 µM and 24 ± 5.2 µM, respectively) compared to BPR and BPL, which aligns with previous studies indicating that electron-withdrawing groups can augment the anticancer efficacy of acridone derivatives. This observation is particularly relevant in the context of drug design, as it highlights the potential for modifying molecular structures to improve therapeutic outcomes.
Molecular docking studies indicated that the synthesized bis-acridone derivatives have favorable binding affinities for topoisomerase I and II, with binding energies ranging from -7.414 to -9.884 kcal/mol. Compound BFM exhibited the highest binding energy of -9.884 kcal/mol, highlighting its strong interaction with the target proteins. These results align with the established function of topoisomerases in DNA replication and cell division, suggesting that bis-acridone derivatives may exert cytotoxic effects by inhibiting these essential enzymes. Various interactions, such as van der Waals forces, ?-? stacking, and hydrogen bonding, help to clarify the binding mechanisms and support the potential of these compounds as topoisomerase inhibitors
The pharmacokinetic profiles predicted through ADMET analysis indicated that the synthesized derivatives possess favorable characteristics, including good bioavailability and acceptable safety profiles. The application of QSAR studies and tools such as Swiss ADME and pkCSM provided a comprehensive understanding of the drug-likeness and toxicity properties of the compounds, which is crucial for advancing these molecules towards clinical applications. The Boiled-Egg model analysis further supported the compounds' potential to traverse the blood-brain barrier, particularly for compound BPL, which is promising for targeting brain tumors.
Moreover, the molecular dynamics simulations provided insights into the stability and conformational dynamics of the bis-acridone derivatives when complexed with topoisomerase II. The observed fluctuations in RMSD and ligand-protein interactions suggest that these compounds engage effectively with their target, which is crucial for their anticancer activity. The identification of key amino acid residues involved in ligand interactions offers valuable information for future modifications aimed at enhancing binding affinity and specificity. study highlights the significant potential of bis-acridone derivatives as anticancer agents, emphasizing the importance of structural modifications in enhancing their therapeutic efficacy. The promising cytotoxicity, favourable pharmacokinetic profiles, and effective binding to topoisomerases position these compounds as viable candidates for further development. Future research efforts should aim to clarify the exact mechanisms of action, investigate potential combination therapies, and conduct in vivo studies to confirm the therapeutic efficacy of these bis-acridone derivatives in cancer treatment. The findings from this study offer valuable insights for further refinement and development of bis-acridone derivatives as promising therapeutic agents for breast cancer and potentially other types of cancer.
CONCLUSION
Bis-acridones synthesis with ethyl chain was a big task because of the steric hindrance with maintaining some parameters successfully synthesized a series of novel N10-substituted bis-acridone derivatives. In-silico molecular docking studies revealed binding energies ranging from -8.28 to -7.557, with compound BFM exhibiting the highest binding energy of -8.11. Cytotoxicity assays using the MTT method on MCF-7 cancer cell lines showed that compound had an IC50 value of 23±4.1 µg/mL. as per HIDE BOILED EGG MODEL the BPL compound cross the Blood Brain Barrier with less toxicity it is helpful for targeted Brain tumor treatment. Comparative analysis of the derivatives suggested that those electron withdrawing groups shows good activity mainly dependent on the chain. As chain increases the activity also increasing with an N10-propyl side chain displayed superior binding affinity and less cytotoxicity, which was further enhanced by the introduction of a fluoro group. The tricyclic ring system and N10-substitution were key structural features contributing to the lipophilicity, binding affinity, and cytotoxic activity. Our findings indicate that the N10-propyl derivative is particularly promising, demonstrating the high binding energies with Topo I and Topo II significant cytotoxic potential. These findings lay the foundation for further refinement and advancement of bis-acridone derivatives as promising anticancer agents.
ACKNOWLEDGMENTS
Authors, thank Poornayu Research Labs, Bangalore for the Proton NMR spectral data, Carbon-13 NMR, and Mass spectra data. The Principal of the Government College of Pharmacy for providing necessary facilities.
Conflict of interest: The authors declare no conflicts of interest in relation to this study.
REFERENCES
(TABLE NO 1) Synthesized compounds and their substitution
|
COMPOUND |
R1 |
R2 |
|
BPL |
H |
H |
|
BPR |
CH3 |
CH3 |
|
BBT |
CH3 |
CH3 |
|
BFM |
CH3 |
F |
(TABLE NO 02): Docking Results
|
DOCKED COMPOUNDS |
TOPO-I (1TL8) |
TOPO-II(3QX3) |
|
|
BPL Bis-acridine-9(10H)-one |
-7.685 |
-8.021 |
|
|
2,7 Methyl BIS |
-4.321 |
-3.912 |
|
|
4,7 METHYL BIS |
-4.076 |
-3.621 |
|
|
PRP PLANE BIS |
-5.456 |
-3.423 |
|
|
BPR 10,10’-(propane-1,3-diyl)bis(2,7-dimethylacridone-9(10H)-one |
-6.194 |
-8.227 |
|
|
3,7 BPR |
-5.304 |
-6.768 |
|
|
4,7 BPR |
-5.208 |
-6.654 |
|
|
BPL PLANE |
-6.031 |
-6.127 |
|
|
BBT 10,10’-(butane-1,4-diyl)bis(2,7-dimethylacridone-9(10H)-one |
-7.194 |
-8.227 |
|
|
3,7 BBT |
-5.289 |
-5.012 |
|
|
4,4 BBT |
-5.345 |
-5.976 |
|
|
PENTA BIS |
-5.876 |
-6.123 |
|
|
PENTA 2,7 BIS |
-6.129 |
-8.456 |
|
|
3,7 BFM |
-6.991 |
-8.021 |
|
|
BFM(10,10’-(propane-1,3-diyl)bis(2-fluro-7-methylacrdine-9(10H)-one) |
-7.414 |
-9.884 |
|
|
3,7 BFM |
-6.991 |
-8.021 |
|
TABLE NO: 3 Inhibition percentages at various treatment concentrations were evaluated.
|
Bis-Compound |
The inhibition percentage was determined against the treated concentrations (µM) |
IC50 (µg/mL) |
||||
|
1 |
5 |
10 |
25 |
50 |
||
|
BPL |
99.93?±?0.11 |
95.52?±?0.96 |
91.63?±?0.45 |
72.81?±?0.20 |
36.05?±?0.81 |
65.53?±?0.58 |
|
BPR |
98.93?±?0.66 |
96.41?±?0.57 |
90.44?±?0.46 |
70.2?±?0.29 |
32.11?±?0.88 |
62.88?±?0.45 |
|
BBT |
96.63?±?0.53 |
94.28?±?0.53 |
89.18?±?0.38 |
68.28?±?0.21 |
24.08?±?0.23 |
52.51?±?0.61 |
|
BFM |
99.76?±?0.56 |
92.09?±?0.42 |
86.1?±?0.88 |
63.35?±?0.19 |
22.78?±?0.69 |
65.13?±?0.44 |
|
DOXORUBICIN |
98.73?±?0.97 |
90.08?±?0.25 |
83.2?±?0.76 |
59.72?±?0.35 |
11.05?±?0.77 |
60.22?±?0.52 |
TABLE NO : 4 Essential characteristics related to physicochemical properties and computational metrics of the compounds examined
|
Bis-Compounds |
BPL |
BPR |
BBT |
BFM |
|
Formula |
C28H20N2O2 |
C33H30N2O2 |
C34H32N2O2 |
C31H24F2N2O2 |
|
MW |
416.47 g/mol |
486.60 |
500.63 |
494.53 g/mol |
|
No. heavy atoms |
32 |
37 |
38 |
37 |
|
No. aromatic heavy atoms |
28 |
28 |
28 |
28 |
|
fraction Csp3 |
0.07 |
0.21 |
0.24 |
0.16 |
|
No rotatable bonds |
3 |
4 |
5 |
4 |
|
No H-bond acceptors |
2 |
2 |
2 |
4 |
|
No. H-bond donors |
0 |
0 |
0 |
0 |
|
Molar Refractivity |
131.84 |
156.51 |
161.32 |
146.49 |
TABLE NO: 5 Analysis of the lipophilic nature of the screened compounds
|
Bis-Compounds |
BPL |
BPR |
BBT |
BFM |
|
TPSA |
2.89 |
57.53 |
74.6 |
46.53 |
|
iLOGP |
3.76 |
0.95 |
1.87 |
3.67 |
|
XLOGP3 |
3.87 |
1.46 |
0.65 |
5.17 |
|
WLOGP |
5.03 |
1.38 |
1.61 |
5.37 |
|
MLOGP |
3.76 |
1.28 |
0.92 |
3.41 |
|
Silicos-IT Log P |
5.23 |
1.22 |
1.85 |
5 |
|
Consensus Log P |
4.33 |
1.26 |
1.38 |
6 |
TABLE NO : 6 Predicted water solubility of the screened compounds
|
Bis-Compounds |
BPL |
BPR |
BBT |
BFM |
|
ESOL log S |
–6.34 |
–8.08 |
–8.31 |
–7.79 |
|
ESOL solubility [mg/ml] |
1.25E–02 |
1.28E+00 |
1.23E+01 |
2.59E–01 |
|
ESOL solubility [mol/l] |
2.657E–05 |
4.65E–03 |
7.48E–02 |
9.87E–05 |
|
ESOL class |
soluble |
moderately soluble |
very soluble |
soluble |
|
Ali log S |
–3.87 |
–4.27 |
–6.79 |
–8.89 |
|
Ali solubility [mg/ml] |
5.19E–03 |
8.73E–01 |
3.68E+00 |
3.74E–04 |
|
Ali solubility [mol/l] |
1.36E–05 |
5.32E–03 |
1.61E–02 |
1.28E–06 |
|
Ali class |
soluble |
soluble |
soluble |
moderately soluble |
|
Silicos-IT log S |
–8.1 |
–7.28 |
–8.56 |
–4.97 |
|
Silicos-IT solubility [mg/ml] |
6.01E–05 |
5.67E+00 |
8.42E+00 |
8.52E–01 |
TABLE NO: 7 Predicted pharmacokinetics (ADME) parameters of the screened compounds
|
Bis-compounds |
BPL |
BPR |
BFM |
BBT |
|
GI absorption |
high |
high |
high |
high |
|
BBB permeant |
yes |
yes |
no |
yes |
|
Pgp substrate |
no |
no |
no |
no |
|
CYP1A2 inhibitor |
no |
no |
no |
no |
|
CYP2C19 inhibitor |
yes |
no |
no |
no |
|
CYP2C9 inhibitor |
yes |
no |
no |
yes |
|
CYP2D6 inhibitor |
no |
no |
no |
yes |
|
CYP3A4 inhibitor |
yes |
no |
no |
no |
|
Log Kp [cm/s] |
–5.87 |
–6.26 |
–7.23 |
–4.41 |
TABLE NO : 8 Predicted drug-likeness, medicinal chemistry and lead-likeness pharmacokinetics parameters of the screened compounds
|
Bis-compounds |
BPL |
BPR |
BFM |
BBT |
|
Lipinski #violations |
0 |
0 |
0 |
0 |
|
Ghose #violations |
0 |
0 |
0 |
0 |
|
Veber #violations |
0 |
0 |
0 |
1 |
|
Egan #violations |
0 |
0 |
0 |
0 |
|
Muegge #violations |
0 |
1 |
0 |
1 |
|
Bioavailability score |
0.55 |
0.56 |
0.56 |
0.56 |
|
PAINS #alerts |
0 |
0 |
0 |
0 |
[Scheme 1] Synthetic Scheme Of Bis-Acridones
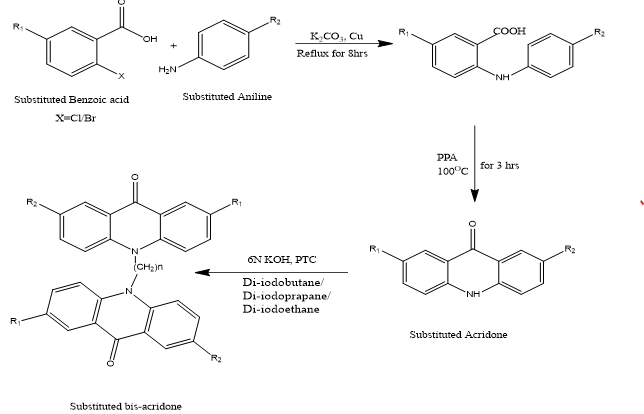
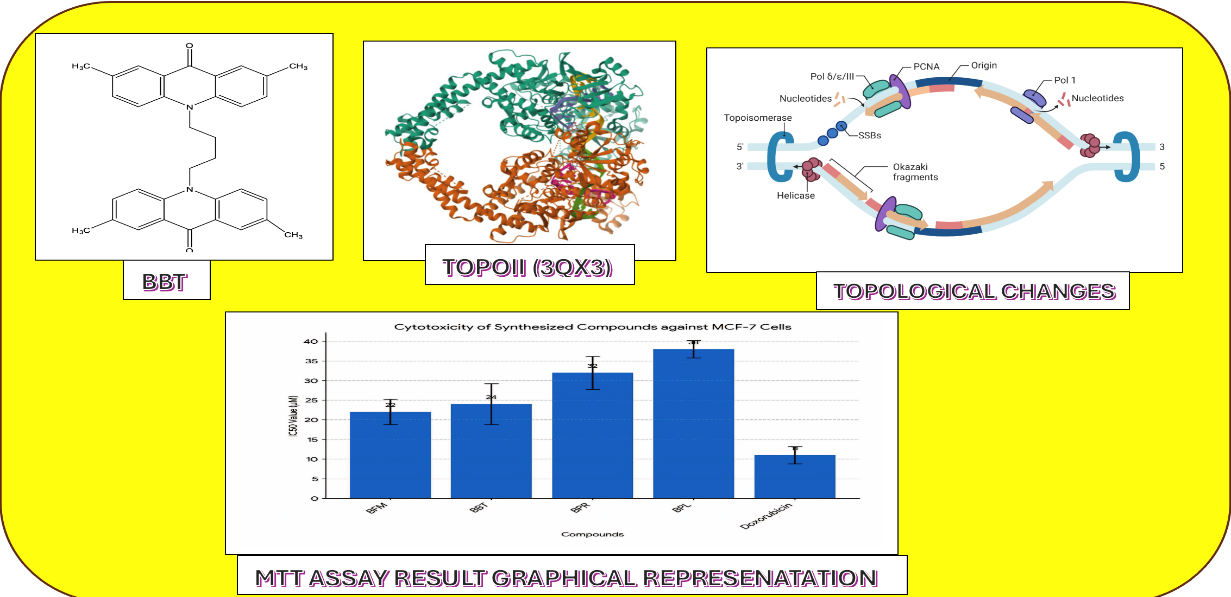
Figure No :1 (Graphical Abstract )
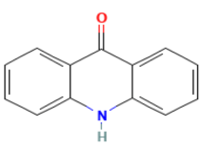
Figure No:2 Structure Of Acridone
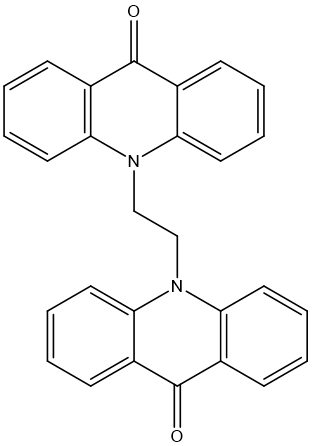
Figure No: 3 Structure Of Bis-Acrdone
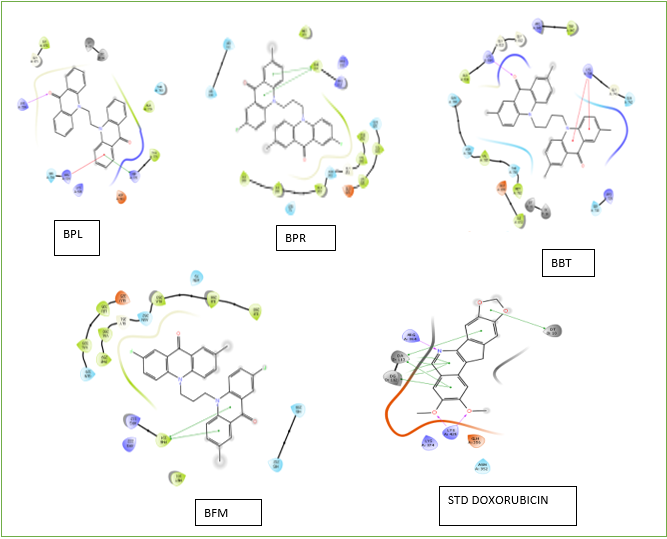
FIGURE 04: Protein-ligand interaction diagram of selected ligands for synthesis and standard PDB ID: 1TL8
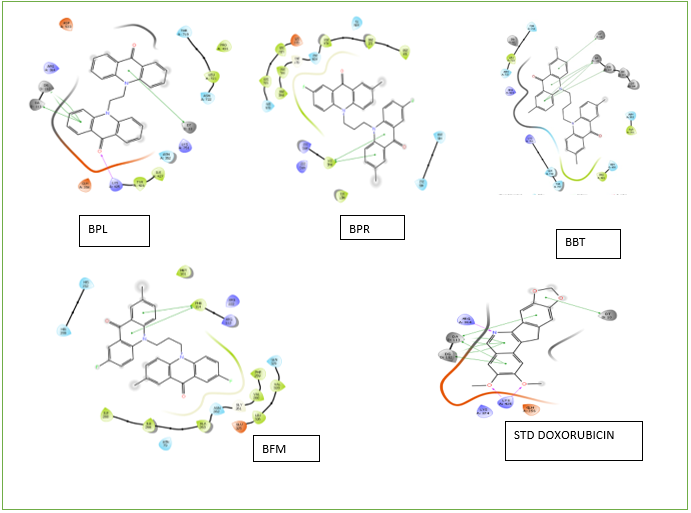
FIGURE 05: Protein-ligand interaction diagram of selected ligands for synthesis and standard PDB ID: 3QX3
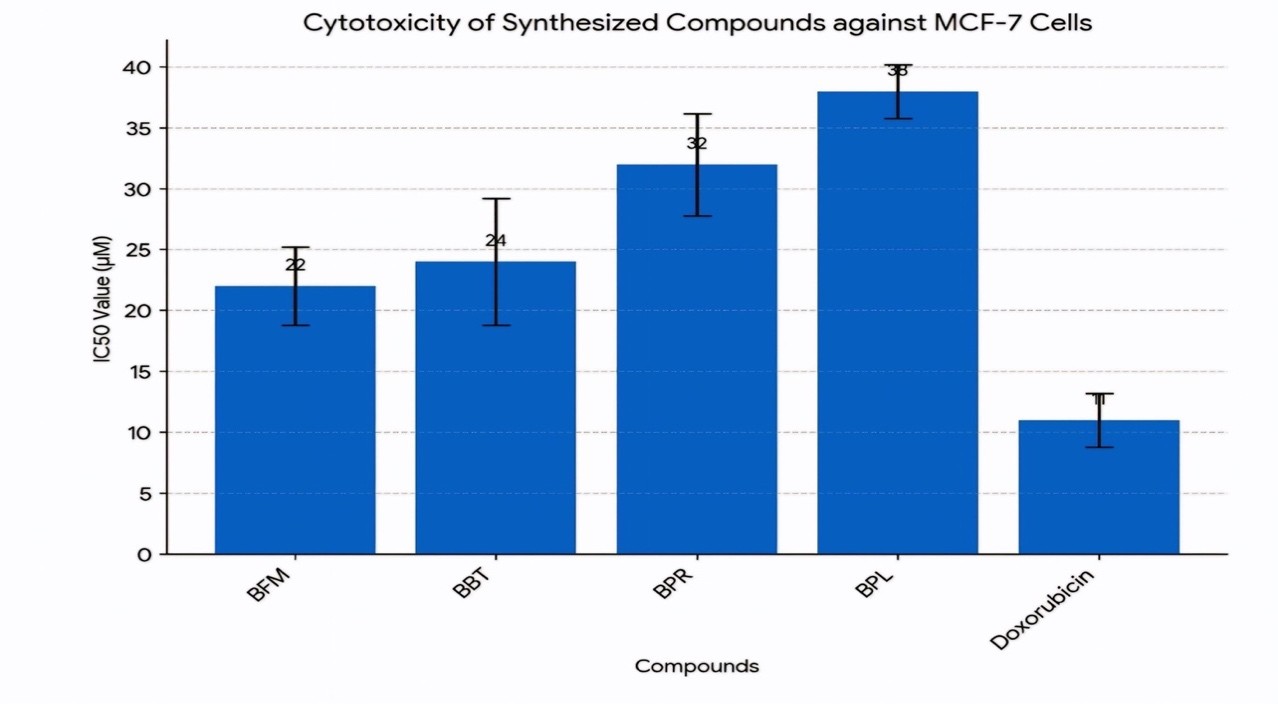
Figure No: 6 Cytotoxicty Graphical Representation
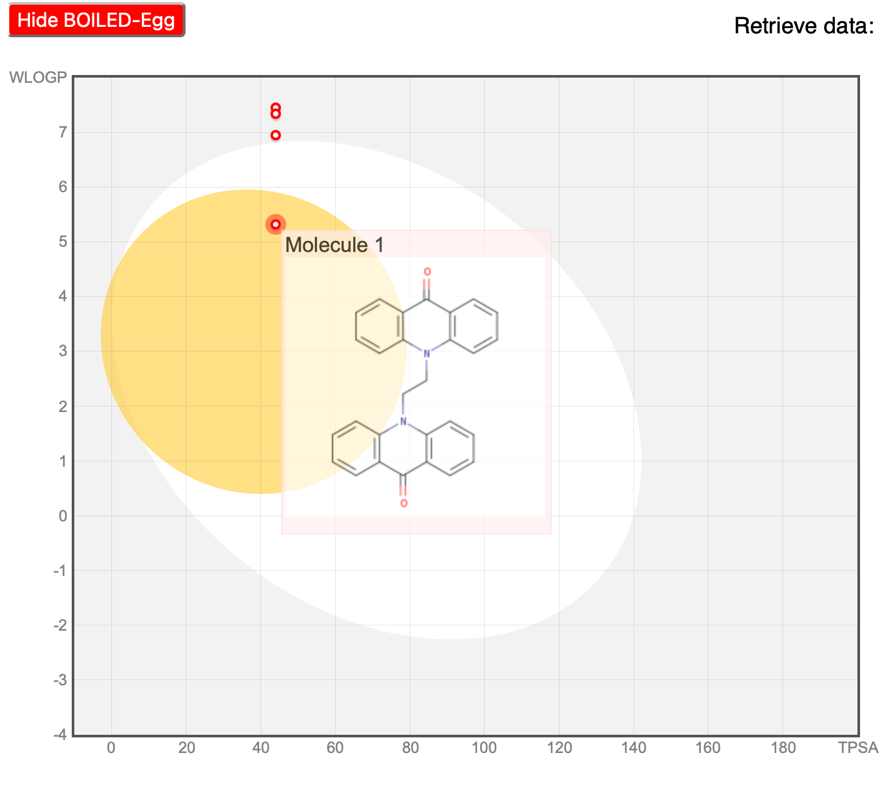
Figure No: 8 Hide Boiled Egg Method
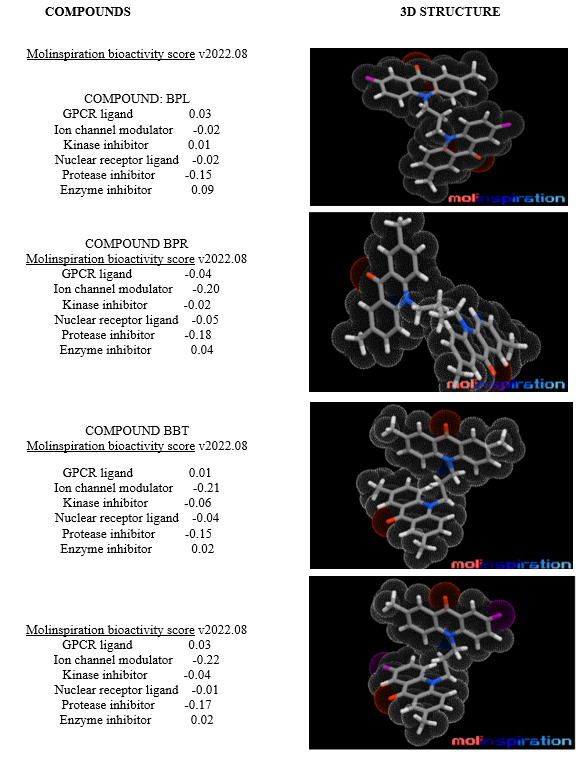
FIGURE NO: 7 Bioactivity and Toxicity Study
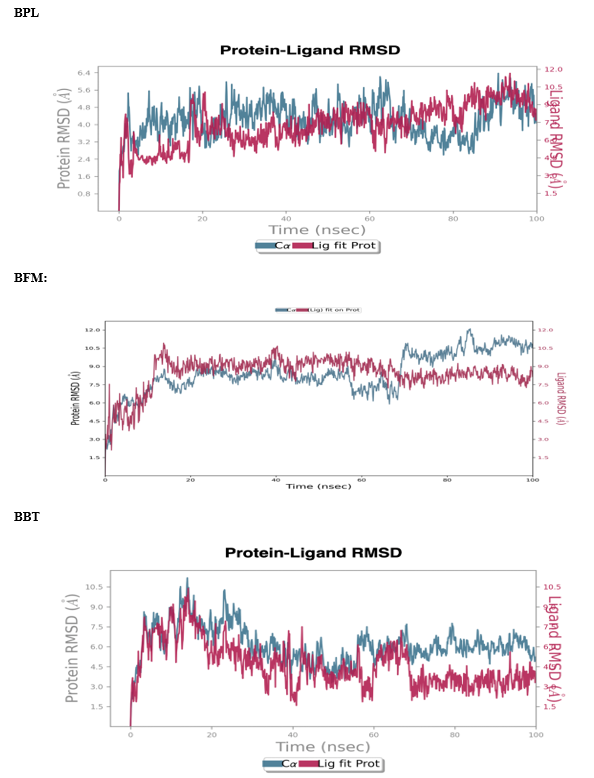
FIGURE NO: 9 Root mean square deviation (RMSD) of simulated complex
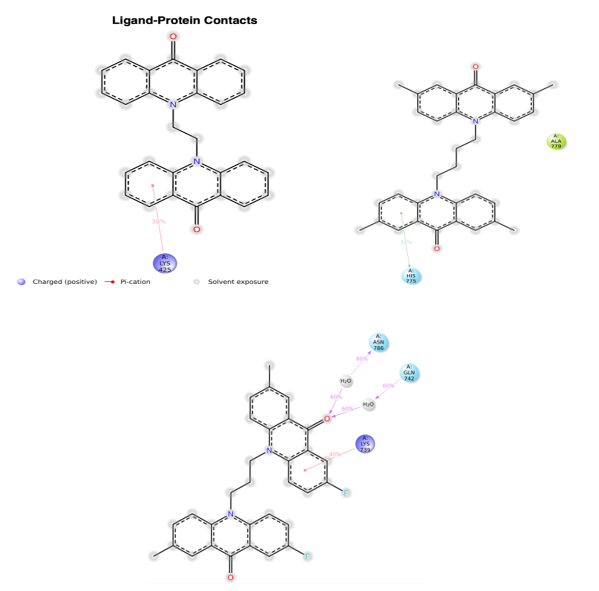
FIGURE NO: 10 ligand-protein interaction probabilities complex during 100 ns time
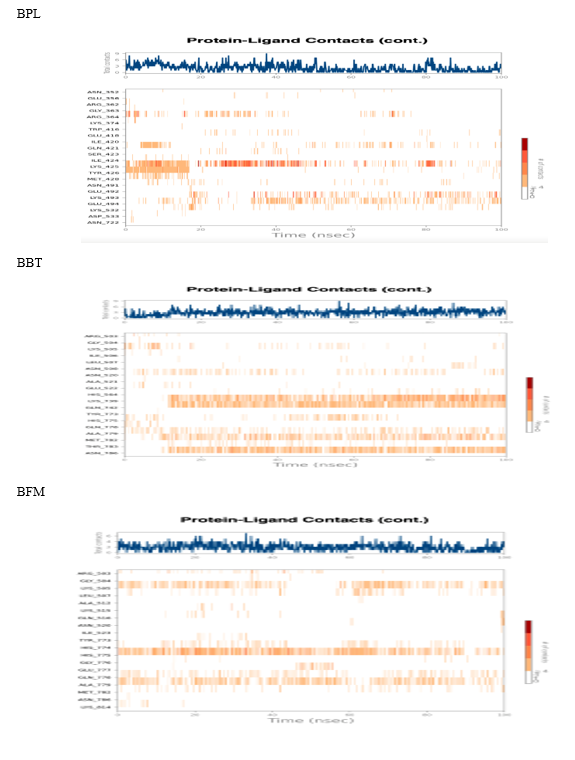
FIGURE NO 11: Protein-ligand interaction diagram for100 ns time period
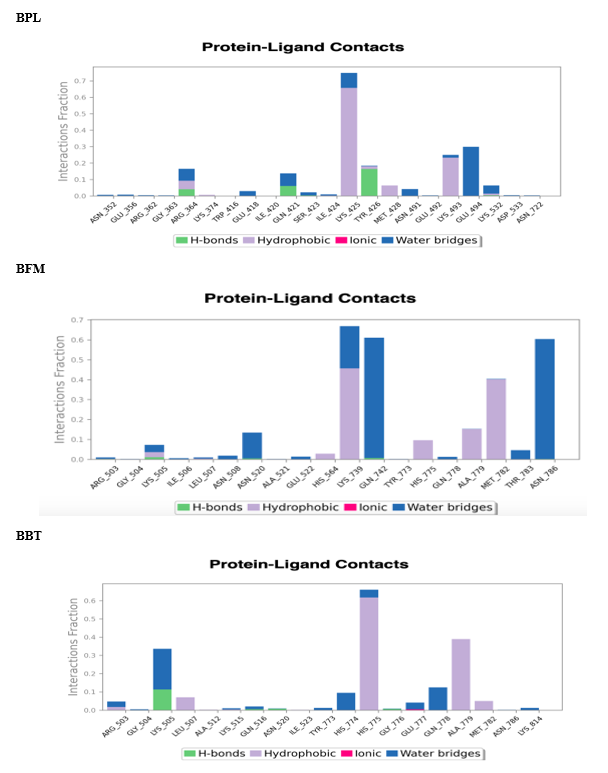
FIGURE NO: 12 Interaction diagrams of complex during molecular dynamic simulations
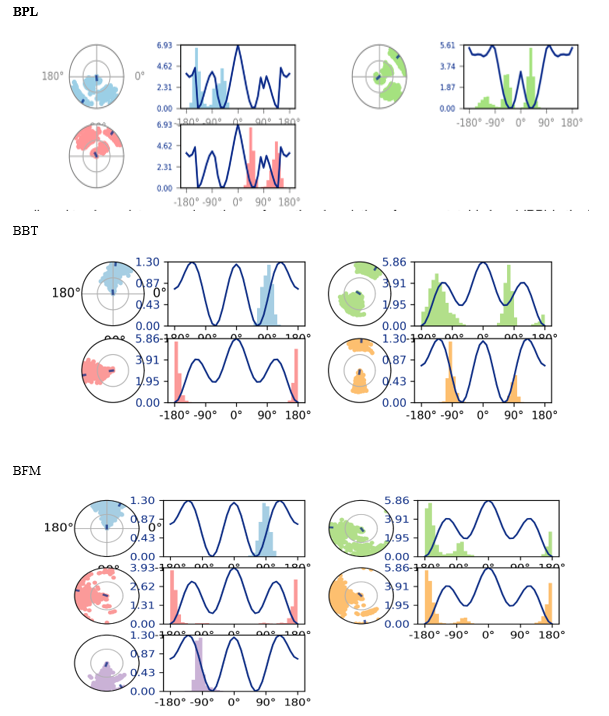
FIGURE NO: 13 Ligand torsion profiles of complex over 100 ns
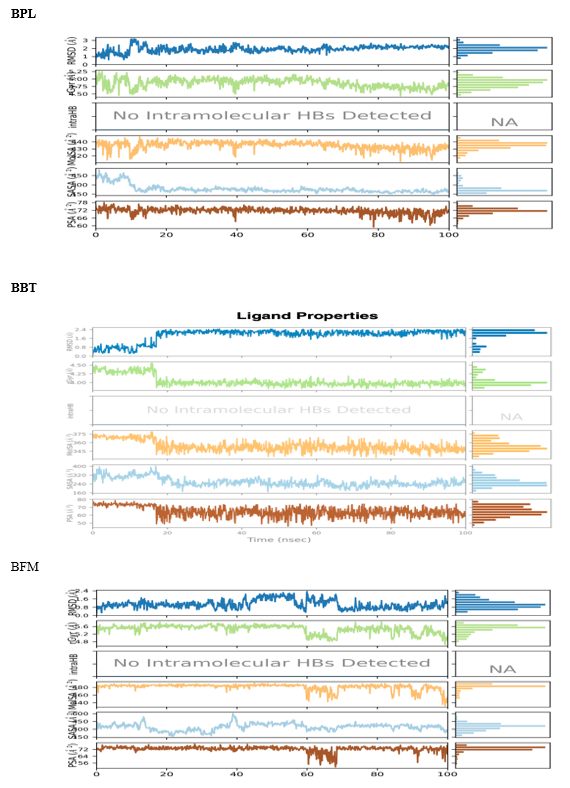
FIGURE NO:14 Fluctuations in the ligand properties over100 ns simulation

Sharanagouda, Dhananjay Mathad, V. V. S. Rajendra Prasad, Jaishree V. A., Bhagyashree Khot, Sathish N. K., Molecular Simulation and Computational Docking Based Synthesis and Screening of Bis-Acridone Analouges, For Antineoplastic Activity, Int. J. of Pharm. Sci., 2024, Vol 2, Issue 12, 1825-1854. https://doi.org/10.5281/zenodo.14325013
 10.5281/zenodo.14325013
10.5281/zenodo.14325013