Degradation studies play a crucial role in the pharmaceutical industry, offering invaluable insights into the stability, safety, and efficacy of drug candidates. This review article provides a comprehensive overview of the methodologies, challenges, and recent advancements in degradation studies aimed at evaluating the stability profiles of pharmaceutical compounds. The review begins by outlining the importance of degradation studies in drug development, emphasizing their significance in predicting the shelf-life and establishing the quality attributes of drug products. Various factors influencing drug degradation, including environmental conditions, chemical properties, and formulation characteristics, are discussed in detail. Moreover, the regulatory considerations governing degradation studies are elucidated, focusing on the guidelines provided by international regulatory agencies such as the International Council for Harmonisation (ICH). Compliance with regulatory requirements is essential to ensure the safety, efficacy, and quality of pharmaceutical products. Finally, the review underscores the significance of interpreting degradation study results in the context of formulation optimization, storage conditions, and manufacturing processes. Strategies for mitigating drug degradation and enhancing stability are proposed, offering practical recommendations for optimizing the development and commercialization of drug candidates. In summary, this review provides a comprehensive understanding of degradation studies for drug candidates, serving as a valuable resource for researchers, pharmaceutical scientists, and regulatory professionals involved in drug development and quality assurance.
Degradation studies, Drug candidates, Stability, Analytical techniques, Regulatory considerations, Formulation optimization.
objective of this review article is to provide guidance for isolating and identifying process related impurities and degradation products from pharmaceutical drug candidates. The identification of degradation products can provide an understanding of impurity formation and define mechanisms. If the identification process is performed at an early stage of drug development, there is adequate time for improvements in the drug substance process and drug product formulation to prevent these impurities and degradants long before the filing stage. Impurity and degradant structure elucidation is a collaborative effort involving the analytical chemist, process chemist and/or formulator as well as the degradation, mass spectrometry and NMR experts. The process described in this two part article uses a designed approach for the impurity and/or degradant identification, which focuses on efficiency so that the success of data collection is maximized. There are number of activities other than collecting experimental data, even though the experiments are central to the process.
The process:-
The process of identification of impurities and/or degradants begins early in drug development. The initial planning and discussion effort can save significant time in experimental stage. A few questions need to be answered at this early stage are:-
- Is this an impurity or degradant problem?
- At what level is the impurity/degradant present?
- Is it a process related impurity (PRI), and if so, at what step of process is it formed?
- Is it a degradant, and if so, under what degradation condition is it formed?
- By gathering all this relevant information, the most efficient method of isolation and identification can be selected.
Purposeful degradation studies:-
- The objective of this section is to provide guidance for developing and executing purposeful degradation experiments for pharmaceutical drug candidates.
- According to ICH guidelines on Impurities in New Drug Products, a degradation product is defined as a chemical change in the drug molecule brought about over time and/or by action of e.g., light, temperature, pH, or water or by reaction with an experiment and/or the immediate container/closure system (also called decomposition product)[4].
- Our goals for this section are to set forth guidelines for these purposeful degradation studies and to describe current procedures for conducting a purposeful degradation study.
- The ICH Guideline on Stability Testing of New Drug Substances and Products provides some guidance on stress testing or purposeful degradation[4]:
- Stress testing helps determine the intrinsic stability of the molecule by establishing degradation pathways in order to identify the likely degradation products and to validate stability indicating power of the analytical procedures used.
- Stress testing conducted to provide data on forced decomposition products and decomposition mechanism. The detailed nature of the studies will depend on individual drug substance and type of drug product.
- The traditional system for developing analytical methods for active pharmaceutical ingredients as well as formulations involves performing purposeful degradation experiments in the method validation phase to challenge the specificity of the methodology[5].
- Using this conventional validation protocol, it is difficult to develop a stability indicating analytical method before having “key stability indicating “ samples generated from purposeful degradation studies to challenge the methodology.
- The 1987 Food and Drug Administration (FDA) Stability Guideline defines stability indicating methodology as[6]:
- Quantitative analytical methods that are based on the characteristic structural, chemical or biological properties of each active ingredient of a drug product and that will distinguish each active ingredient from its degradation products so that the active ingredient content can be accurately measured
- Purposeful degradation studies are not one time event. They are initiated in the introductory phase of the development process and continue over the lifetime of a candidate. Purposeful degradation studies include appropriate solid and solution state challenges to mimic potential future storage conditions including acid/base hydrolysis, thermal/humidity, oxidation, and light exposure in accordance with ICH guidelines. For drug product studies, placebo and drug substance are run as control samples for each condition.
- Under this approach the purposeful degradation protocol is based on the drug substances and drug product chemistry. The analytical methodology is challenged with sample degraded using “reasonable condition”. The definition of reasonable conditions is 10-20?gradation of the pharmaceutical active ingredient.
- Kinetic points are obtained for each condition along the degradation pathway to gain a better understanding of mechanism. Using this approach analyst can focus on the key primary degradation products and can generate degradation samples more predictive of ICH stability.
- The ICH guideline on stability testing on New Drug Substance and Products states the following for stress testing of purposeful degradation[4]:
- It is recognised that some degradation pathways can be complex and that under forcing conditions degraded products may be observed which are unlikely to be formed under accelerated or long term testing. This information may be useful in developing and validating suitable analytical methods, but it may not always be necessary to examine specifically for all degradation products.
- Degradation samples are analysed at the initial phases of high performance liquid chromatography (HPLC) method development using purity (for area percent values of degradants) and potency methods (to obtain assay values for the amount of active ingredient remaining against reference standards).
- By performing those analyses, the analyst is able to obtain mass balance data and a better understanding of the level of degradation occurring.
- For all these conditions, samples can be examined for any changes in physical properties (appearance, clarity, and colour). If polymorphism is concern, then degradation samples can be analysed for polymorphic conversion[7,8].
- For color measurements, a useful degradation instrument is a colorimeter. Unlike the human eye that cannot quantify color accurately, a colorimeter expresses colors numerically according to international standards[9].
- LAB color space is presently one of the most popular and uniform color spaces.L, A, and B refer to the three axes of the system: a lightness axis (L) and two axes representing both hue and chroma, red-green (A) and blue-yellow (B)[9].
4. Regulatory Requirements:-
From a regulatory perspective, forced degradation studies provide data to support the following:-
- Identification of possible degradants,
- Degradation pathways and intrinsic stability of the drug molecule,
- Validation of stability indicating analytical procedures.
Issues addressed in regulatory guidances include:-
- Forced degradation studies are typically carried out using one batch of material.
- Forced degradation conditions are more severe than accelerated stability testing such as > 50°C; ?75% relative humidity; in excess of ICH light conditions; high and low pH, oxidation, etc.
- Photostability should be an integral part of forced degradation study design.
- Degradation products that do not form in accelerated or long term stability may not have to be isolated or have their structure determined.
- Mass balance should be considered.
Issues not specifically addressed in regulatory guidance:-
- Exact experimental conditions for forced degradation studies (temperature, duration, extent of degradation, etc.) are not specified.
- Experimental design is left to the applicant’s direction.
5. Degradation Prediction Tools:-
-
- CAMEO is a computer program that predicts the products of organic reactions given starting materials, reagents and conditio. The analyses cover the following key degradation conditions: basic/nucleophilic, acidic/electrophilic, radical, oxidative/ reductive and photochemical as well as mechanistic interpretations of these reactions.
- In general, the CAMEO algorithms have been designed to give product mixtures that err on predicting more degradation products than actually observed. This is preferable to rules that are too restrictive and reject a key product observed in actual degradation or ICH stability studies. It is also likely that certain products predicted can undergo further decomposition.
- Due to these limitations with this prediction program, tracking historical degradation data in terms of functional groups along with CAMEO prediction data provides a more thorough approach to degradation prediction exercises.

Fig. 1. Forced degradation process flow map—prediction to documentation in a structure searchable global degradation database.
6. Drug Substance Degradation Studies:-
- Purposeful degradation studies of the drug substance include appropriate solution and solid state stress conditions (e.g; acid/base hydrolysis, heat, humidity, oxidation, light exposure, in accordance with the ICH guidelines). The ICH guideline specifically state:-
- Stress testing is likely to be carried out on a single batch of material and to include the effect of temperature in 10°C increments above the accelerated temperature test condition (e.g., 50°C, 60°C, etc); humidity where appropriate (e.g. 75%RH or greater); oxidation and photolysis on the drug substance plus its susceptibility to hydrolysis across a wide range of pH values when in solution or suspension.
- The analytical methods developed need to separate degradants observed on stability; therefore, it is critical that the stress testing model be realistic. Excessive stress will lead to decomposition beyond primary degradation products. This level of stress will cause unnecessary method development for separation of components that will never be observed upon storage according to ICH guidelines.
- The specific conditions (intensity and length of time) will depend on the chemical characteristics of the drug substance.
- If there are multiple salt forms or polymorphs being developed in parallel, is helpful to perform comparative purposeful degradation studies on each form. If there are stability issues with the salt form, analysis of the salt without drug substance become advantageous step.
- In terms of quantity of material, the recommended amount needed for experimental purpose is at least 300 mg of drug substance at the early stage of development if there are limited quantities of material and in the case of no limitation of material, there can be needed 10-15gm of drug substance at the last stage of development.
- All samples generated should be stored or at below 5°C to preserve kinetic points until HPLC screening can be performed.
- Acid/Base Stress Testing:-
- Acid/Base stress testing is performed to force the degradation of a drug substance to its primary degradation products by exposure to acidic and basic conditions over time. Functional group likely to introduce acid/base hydrolysis are amides(lactams), esters(lactones), carbamates, imides, imines, alcohols(epimerization for chiral centers), and aryl amines.
- To initiate acid/base studies, a preliminary solubility screen of the drug substance is performed. Solubility of at least 1mg/ml in 1N acidic and 1N basic conditions is recommended for the acid/base stress testing; however; concentration less than 1 mg/ml can be used if solubility is an issue. In some cases, a co-solvent may be necessary to achieve the target concentration.
- For Acid:-
- Example acids include HCL or H2SO4 (0.1-1 mol/L solution). Studies should be carried out in the solution state.
- For certain APIs that are partially soluble or insoluble in the described acidic solution, addition of an appropriate co-solvent, or adjustment of solution pH in the acidic range may be required to achieve dissolution; or the APIs can be run as suspensions Special attention to the API structure should be paid when choosing the appropriate co-solvent (i.e. do not use alcohols for acidic conditions due to their reactivity).
- Dimethylsulfoxide, acetic acid and propionic acid are useful under acidic conditions. Additionally, the sample may be heated for a defined time/temperature to accelerate degradation, depending on the API sensitivity to heat.
- For Basic:-
- Example bases include NaOH, LiOH or KOH (0.1–1 mol/L solution).
- Studies should be carried out in the solution state. For certain APIs which are partially soluble or insoluble in the described basic solution, addition of an appropriate co-solvent, or adjustment of solution pH may be required to achieve dissolution; or the APIs can be run as suspensions.
- Glyme and 1, 4-dioxane facilitate reactions in basic conditions . Additionally, the sample may be heated for a defined time/temperature to accelerate degradation, depending on the API sensitivity to heat.
- A stock solution of the drug substance at the appropriate concentration using water and co-solvent (if necessary) is prepared for the set of acid/base hydrolysis studies. This solution is used to prepare the acid and base solutions as well as the drug substance “as is” control. Different co-solvents can be used in the acid and base degradation studies. If the co-solvent is not the same, drug substance “as is” and acid/base control (no drug substance) samples will need to be prepared in each of the solvent systems.
- By using this acid/base technique, samples can be monitored on-line without the need to quench before HPLC analysis.
- Kinetic points taken along the reaction pathway are stored at or below 5 ?C until HPLC screening is conducted. Often the acid/base reactions proceed too rapidly even at low temperature and a quenching step may be necessary to hold a critical kinetic product distribution.
- If a quenching step is required, a generic quenching sequence involves the use of 1 equivalent of the conjugate acid or base followed by 2.5 equivalents of ammonium acetate for neutralization. One of the major disadvantages of the quenching step is solubility.
- If the drug substance is very insoluble at neutral pH, the drug substance may precipitate out of solution in the neutralization step, and as a result, further solubility experiments may be necessary. Therefore, the technique of choice involves on-line HPLC screening of acid/base hydrolysis samples without pH adjustment and only temperature.
- cooling of samples to preserve the kinetic product distribution. Another critical parameter in the acid/base hydrolysis experiment involves incorporation of the appropriate controls.
- The critical controls are “as is” drug substance and acid only and base only at an appropriate concentration without the drug substance. Additionally, if elevated temperature is required, a thermal control sample should be run at the reaction temperature without acid or base to determine whether the degradation observed is the result of thermal or acid/base degradation. If acid/base quenching is involved, the controls should also be quenched.
- The acid and base stress conditions should result in approximately 10-20?gradation of the drug substance or represent a reasonable maximum condition achievable. If this level of degradation is not achieved, additional hydrolysis experiments should be performed at no more than 70 ?C for a 1-week total reaction time. Going above this level of stress is not recommended for typical drug substance materials.
- Excessive acid/base stress will produce nonpredictive samples and will lead to unnecessary effort in the HPLC method development.

TABLE:- 1. Guidance on Acid/Base Experimental Setup
- Thermal and Thermal/humidity Stress Testing:-
- Solid state stability can be evaluated utilizing accelerated storage temperatures in general greater than 50 °C and N75% relative humidity. The duration of exposure is dependent on the API sensitivity.
- If the forced degradation thermal/humidity conditions produce a phase change, it is recommended to also run thermal/humidity conditions below the critical thermal/humidity that produces the phase change.
- The goal of thermal and thermal/humidity studies is to force the degradation of drug substances over time to determine the primary thermal and/or humidity degradation products.
- To evaluate stability utilizing elevated temperatures (above the ICH guidelines for accelerated thermal humidity challenges), stress conditions are selected based on a conservative estimate of the Arrhenius expression—a quantitative relationship of reaction rate and temperature using an average activation energy.
- Arrhenius kinetics may be used to establish an appropriate temperature and maximum duration of thermal degradation studies. Using an appropriate assumption of activation energy, the duration of controlled room temperature storage that is simulated by the study can be estimated.
- Based on this estimate, a 10°C increase in temperature results in a doubling of the reaction rate and a decrease in the reaction time by a factor of 2.
- Using this rule of thumb, 1 year at 30°C is equivalent to 3 weeks at 70°C. As a result, the recommended study length for samples to predict a 2-year room temperature shelf life is 6 weeks at 70°C.
- This estimate is more likely to be true at or near room temperature. It is also worth noting that increasing the energy of the system may produce products not seen under ICH stability guidelines because there is more energy available to reach activation barriers to products that cannot be formed under ICH conditions.
- In these cases, it is essential to compare results to ICH stability data and run experiments over a few temperature increments to get a better correlation to ICH stability.
- Additionally, it is important to determine early kinetic points at 70 °C to get an understanding of the primary degradants.
Kobs = Aexp (-Ea/RT)
- Where, kobs is a specific rate constant, A is the preexponential factor, Ea is activation energy, T is temperature in degrees Kelvin, and R is the gas constant (1.987 cal K-1 mol-1).
- To perform a 70 ?C challenge, an example kinetic pull point setup is executed over a 6-week time frame with more pulls early in the reaction time scale.
- For thermally unstable compounds, the early kinetic points enable the analyst to observe the primary degradation products that may have further converted by the end of the 6-week study.
- Humidity stress is also factored into this model. The starting stress conditions with humidity levels are 70°C/30% RH (ambient humidity) and 70 °C/75% RH.
- These starting conditions are modified if a compound undergoes a change in physical form at or below this elevated temperature.
- In these cases, significant non-Arrhenius behaviour can be observed. Additionally, the stress testing temperatures are significantly reduced if the pharmaceutical candidate has a low temperature storage recommendation (for example, 5 °C). In this scenario, a more reasonable stress level would be 40 °C.

TABLE:- 2 Guidance for Thermal Stress Length of Study
- It is critical to ensure the integrity of the solid samples throughout the study. For example, deliquescence under high humidity conditions is often observed.
- To detect percent water gain/loss, it is useful to perform gravimetric analysis on all degradation samples.
- Other methods of water analysis such as Karl Fischer titrimetric determination can also be used (see Section I.B). These methods of analysis can help determine if water has an effect on the degradation mechanism.
- When these thermal degradation experiments are performed,the neck of each flask should be filled with a cotton plug to avoid problems with condensation entering the vial.
- The cotton plug is added loosely to allow passage of air into and out of the vial, but tight enough to avoid loss of sample should the vial be accidentally inverted. Water gain/loss data and physical appearance data are key tools for early prediction of formulation stability issues upon ICH stability storage.
- According to the thermal/humidity stress testing conditions selected, samples are placed into appropriate ovens. If humidity ovens capable of 70 ?C/30% RH and 70 ?C/75% RH are not available, saturated salt solutions contained in desiccators can be used to control humidity accurately.
- These conditions are particularly useful for high-potency drug substance compounds for which samples must be contained. A saturated NaCl solution is used to obtain conditions of 75% RH at 70 °C and a saturated MgCl2 solution is used to obtain conditions of 30% RH at 70 °C

TABLE:- 3 Guidance for Thermal/Humidity Experimental Setup
Oxidation:-
- Oxidation can be carried out under an oxygen atmosphere or in the presence of peroxides. The use of oxygen is a more realistic model. Free radical initiators may be used to accelerate oxidation.
- Generally, a free radical initiator and peroxide will produce all primary oxidation degradation products observed on real-time stability. Therefore, free radical and/or hydrogen peroxide conditions are strongly recommended at all stages of development.
- For solution state stress conditions, dissolve the API utilizing an appropriate solvent, add 5–20 mol% of a free radical initiator at atmospheric pressure. To increase the solubility of oxygen in the solution, the reaction can be performed in a reaction vessel pressurized at 50–300 psi with molecular oxygen.
- Additionally, the system is heated to accelerate degradation. The temperature depends on the free radical initiator selected.
- For peroxide conditions, hydrogen peroxide reagent (up to 3%) can be used. As previously indicated, the addition of an appropriate co-solvent may be necessary, depending on API solubility.
- Hydrogen peroxide stress testing can be useful in DP studies where hydrogen peroxide is an impurity in an excipient. Solid-state stress conditions may be similarly investigated by placing the API (as is) in suitable closed containers filled with an oxygen headspace versus an argon or nitrogen control headspace.
- Additionally, the sample may be heated for a defined time/ temperature to accelerate degradation, depending on the API sensitivity to heat. For later stage development compounds when more time and effort can be focused on mechanistic understanding, the following oxidation conditions can be applied.
- The addition of metal ions to solutions of API can indicate whether there is a tendency for the API to be catalytically oxidized. Iron and copper ions are routinely found in APIs and formulation excipients.
- Transition metal ions can also reduce peroxide to generate hydroxyl radicals in a Fenton-type reaction. In addition, light can also effect oxidation reactions. Light absorbed by a photosensitizer can react with molecular oxygen to form the more reactive singlet oxygen species[10].
- To prepare for the oxidative degradation study, a preliminary solubility analysis of the drug substance should be performed. The reaction solvent can mimic the proposed formulation.
- For example, if the formulation has a protic environment, a protic solvent can be selected whereas an aprotic solvent can be selected for an aprotic formulation environment.
- Acetonitrile is the preferred solvent over methanol because alcohols slow the reaction by competing with the drug for initiator radicals[14].
- Oxidative purposeful degradation studies typically require solubility of approximately 1–10 mg/mL in unbuffered conditions to achieve reasonable levels of degradation.
- The maximum concentration in this range is recommended since the free radical oxidation process is concentration dependent. A co-solvent may be necessary to achieve the target concentration range. Our pressurized oxidation approach is run with radical initiators to accelerate oxidation.
- The process involves a radical chain reaction between molecular oxygen and the pharmaceutical drug candidate, a process known as autoxidation[15-17].
- Key predictive samples with 10–20?gradation are typically generated within 10 days using the addition of 1–10 mole % radical initiator.
- The free radical process of autoxidation consists of a chain sequence involving three distinct types of reactions: initiation, propagation, and termination. The initiation produces a free radical to begin the chain reaction[18].
- Use of a radical chain initiator is a valid method of accelerating autoxidation.
- Above table shows example diazenes one can use to accelerate the pressured oxidation reactions for aqueous and organic based systems.
- The radical chain initiating diazene undergoes thermal bond homolysis to yield two radicals and molecular nitrogen. A hydrogen atom is abstracted from the drug substance or excipient.
- Radical addition occurs in the initial propagation step of autoxidation. Molecular oxygen then reacts with the drug substance or excipient free radical at the diffusion-controlled rate of approximately 109 M-1s-1 depending on the solvent system.
- The propagation sequence continues as a peroxyl radical abstracts a hydrogen atom from the drug substance or other organic substrate present such as an excipient in the case of drug product.
- This rate-determining step, which has a rate constant referred to as kp, generates a second free radical to propagate the chain and yield a hydroperoxide product. Under these conditions, the rate of oxidation is given by equation, where kp and kt are the propagation and termination rate constants, respectively.
- Ri is the rate of initiation and R ? H is the substrate. Termination of the autoxidation process occurs as peroxyl radicals couple to produce nonradical products. Additional sources of free radicals to initiate the free radical chain process include ultraviolet (UV) light and heavy metals (copper, iron, cobalt, manganese, and nickel) which catalyze oxidation by shortening the induction period and promoting free radical formation[19].
-dO2/dt = kp [R-H] (Ri1/2/2kt1/2)
- To perform an oxidative degradation study, the drug substance is dissolved in an appropriate solvent and transferred to a reaction vessel pressurized at 50– 300 psi with molecular oxygen to increase the oxygen solubility in solution.
- Additionally, the system is heated to accelerate degradation. Oxygen solubility depends on the solvent used in an oxidation reaction. For example, at 0 ?C the concentration of oxygen in methanol at atmospheric pressure is 11 times the concentration of oxygen in water.
- As the temperature is increased to 40 ?C, the oxygen solubility in methanol is 21 times greater than that in water[20,21].
- Kinetic points are taken along the reaction pathway and quenched using 1–10 mole%antioxidant. Antioxidants work by consuming oxygen at a faster rate than the drug substance and can compete for free radicals (thus, they are termed free radical scavengers).
- These antioxidants will protect the drug substance and/or excipients until the free radicals are consumed.
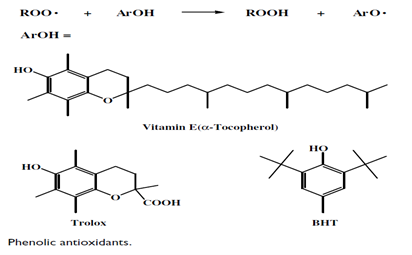
- The most common antioxidants are ascorbic acid, butylated hydroxyanisole, butylated hydroxytoluene (BHT), and sodium sulphite[22,23].
- The phenolic antioxidants readily react with peroxyl radicals to generate a hydroperoxide product and relatively unreactive radical.
- Thus, phenolic antioxidants are also excellent hydrogen atom donors. Ascorbic acid and trolox can be used for aqueous based systems, whereas BHT and vitamin E can be used for oxidation performed in organic solvents[24-26].
- The most effective chain-breaking antioxidant among the phenolic antioxidants is ?-tocopherol, or vitamin E. Vitamin E is an organic soluble phenolic antioxidant. For aqueous systems, trolox is a better choice for a phenolic antioxidant[27].
- Because of its chromanol structure, it has effective antioxidant activity and its carboxyl group provides moderate water solubility[24].
- In systems where metal catalysis of oxidation is an issue, the use of a metal chelator such as ethylenediaminetetraacetic acid is recommended to inhibit oxidation.
- Additionally, as with previous degradation techniques, the oxidation samples should be stored at low temperature (below 5 ?C) to preserve the primary oxidative products and prevent them from decomposing further.
- This will enable continued analysis of the primary oxidative degradants during the method optimization phase of development. It is critical to run the appropriate controls.
- A thorough experimental design should incorporate the following controls containing the drug substance: oxygen with initiator, oxygen without initiator, argon or nitrogen with initiator (purged of oxygen), and a thermal control (at the reaction temperature).
- Incorporating the samples outlined in Table will allow easier interpretation of the degradation results and enable one to get a better mechanistic understanding of whether the degradation results from thermal, free radical, or nonfree radical -processes.
- The initiator and antioxidant at appropriate levels without drug substance are other essential controls to run in a method set to determine if any peaks shown in the analytical method result from oxidation reaction compounds that are not drug substance related.

TABLE:- 4 Guidance for Oxidative Degradation Experimental Setup
Photostability:-
- The goal of the photostability studies is to force the degradation of drug substances via UV and fluorescent conditions over time to determine the primary degradation products.
- UV and visible light are the most energetic electromagnetic radiation sources to which pharmaceutical drug substances and drug products are typically exposed.
- A molecule absorbs light when an absorption band exists that overlaps to some extent with the incident light energy and a valence electron in the relevant chromophore is raised to an excited state[28].
- Light stress conditions can also induce photooxidation by free radical mechanisms. The photo-excited molecule undergoes dissociation and the free radical formed is scavenged by molecular oxygen.
- The resultant peroxy free radical may then undergo reactions yielding a mixture of oxidized products.
- Functional groups likely to introduce drug photoreactivity are as follows: carbonyl, nitroaromatic, N-oxide, alkenes, aryl chlorides, weak C H and O H bonds, sulfides, and polyenes[29].
- There are two types of studies used in pharmaceutical photostability testing: stress testing (purposeful degradation) and confirmatory testing. Purposeful degradation is used to evaluate the overall photosensitivity of the material (unprotected drug substance and drug product) for method development purposes and/or degradation pathway elucidation.
- Confirmatory testsare used to determine if there is a need to protect the final product from light. The ICH Guideline on Stability Testing of New Drug Substances and Products notes that photostability should be an integral part of stress testing[30].
- The only requirements are that the source(s) be continuous over the near UV and visible spectral regions.
Option 1:
Any light source that is designed to produce an output similar to the D65/ID65 emission standard such as an artificial daylight fluorescent lamp, combining visible and ultraviolet (UV) outputs, xenon lamp, or metal halide lamp. D65 is the internationally recognized standard for outdoor daylight as defined in ISO 10977 (1993). ID65 is the equivalent indoor indirect daylight standard. For a light source emitting significant radiation below 320 nm, an appropriate filter(s) may be fitted to eliminate such radiation.
Option 2:
For Option 2, the same sample should be exposed to both the cool white fluorescent and near ultraviolet lamp. A cool white fluorescent lamp designed to produce an output similar to that specified in ISO 10977 (1993); and a near UV fluorescent lamp having a spectral distribution from 320 nm to 400 nm with a maximum energy emission between 350 nm and 370 nm; a significant portion of the UV should be in both bands of 320 to 360 nm and 360 to 400 nm. The sample must be exposed to both sources however, the exposure can be in a sequential or simultaneous set-up[31,32].
Drug Product Degradation Studies:-
- Drug product (DP) degradation cannot be predicted solely from the stability studies of the API in the solid state or solution.
- The non-active pharmaceutical ingredients can also react with the API or catalyze degradation reactions. Impurities in the excipients can also lead to degradation in the DP not originally observed in the API.
- For DP formulations, heat, light, and humidity are often used. The DP stress conditions should result in approximately 5–20?gradation of the API or represent a reasonable maximum condition achievable for a given formulation.
- The specific conditions used will depend on the chemical characteristics of the DP. For a solid DP, key experiments are thermal, humidity, photostability and oxidation, if applicable. For solution formulations, key experiments are thermal, acid/ base hydrolysis, oxidation and photostability.
- It is recommended to compare stressed samples with unstressed samples and an appropriate blank.
- For DP studies, the blank sample is an appropriate placebo. The stressed placebo sample will provide information about excipient compatibility. It is advised to take kinetic time points along the reaction pathway for API and DP degradation studies to determine primary degradants and a better understanding of the degradation pathway.
- For example, when a formulation is exposed to light, other compounds such as excipients or impurities absorb light to become excited and transfer energy to the drug molecule, leading to photoreaction. Purposeful degradation studies are performed to determine the physical and chemical compatibility of the drug substance with excipients. These studies on the drug product depend on the chemical composition of the drug product formulation.
- The drug product stress conditions should result in approximately 10–20?gradation of the active drug substance or represent a reasonable maximum condition achievable for a given formulation.
- The specific conditions (intensity and length of time) used will depend on the chemical characteristics of the drug product. For all drug product studies, it is critical to run the proper controls: the drug substance, drug product, and placebo. For the most complete understanding of the degradation pathway, all three samples should be taken at each kinetic point and analyzed by the pharmaceutical drug candidate HPLC screening method.
- In the chromatographic screening of degradation samples, it is extremely useful to use the same methods for drug substance and drug product to allow easier understanding of chromatographic differences.
- For drug product, the following key experiments should be considered. These experiments will vary depending on whether the formulation is a solution or solid drug product.
- For a solid drug product, key experiments are thermal, humidity, photostability (in accordance with ICH guidelines), and oxidation, if applicable. The most common type of interaction in solid dosage forms is between water and the drug substance. Hence, thermal and thermal/humidity challenges are critical.
- For solution formulations, key experiments are thermal, acid/base hydrolysis, oxidation and photostability (in accordance with ICH conditions).
- For a solution drug product, more emphasis should be placed on acid/base hydrolysis. A design involving the drug product typically adjusted ±1 or 2 pH units around the target pH is acceptable. In addition, earlier time points are required for solution light stability due to the increased rate of reaction in the solution phase.
- These earlier kinetic time points are generally required in the range of 0–1× ICH for photostability.
- Unlike solids and semisolids, stability of drugs in solution can be better predicted and basic kinetic studies can be applied.
- Semisolids (as oils) are more susceptible to oxidation due to autoxidation of the oil excipients; hence, for these formulations, oxidation is a critical experiment. Drug products that are liquids should be stressed in chemically inert containers.
- Additionally, effects of actual drug product storage containers should be built into the solution drug product studies since the container is considered to be a part of the formulation.
- Oxygen permeation is also more significant in plastic than in glass containers; hence, the container should be considered in the drug product design as well.
- In terms of the amount of drug product to be set up, a later-stage study requires approximately 100 tablets (solid) or ?1 L (solution). Early-stagework requires approximately 25 tablets (solid) or ?50 mL (solution). As outlined in Section II.B, the typical length of a study is approximately 6 weeks.
- This can be reduced by a factor of 2 for early stage work (phase I clinical trials) where timelines can be extremely tight.
- A final optimized formulation is not necessary to begin purposeful degradation studies. Timing for the final optimized formulation may be too late in development to fully understand the degradation.
- Early understanding of degradation can be fed back to the formulators for improvement in the formulation before an ICH stability study is well under way and a stability problem is detected.
- The formulator typically has determined excipients early and is finetuning ranges. The purposeful degradation experiments use a formulation that gives the maximum possible excipient/active interactions. This is a designed approach based on the chemical characteristic of the drug substance/drug product. All excipient chemical reactions should be incorporated into the experimentaldesign.
- For example, drugs that contain primary and secondary amines functionality undergo Maillard reactions with lactose and other reducing carbohydrates such as glucose and maltose under pharmaceutically “reasonable conditions.”
- This reaction should be considered during formulation development. Alternative excipients such as mannitol, sucrose, and trahalose, which are not subject to the Maillard reaction, should be used in place of lactose in such cases.
- It is key to integrate drug substance and drug product degradation studies, with the drug substance being a critical control for drug product degradation studies. In particular, this aids in the determination of whether a degradant is related to the drug substance or to an excipient.
- Another point worth noting is to use the same lot of drug substance that is present in the drug product.
- This minimizes potential variables that can complicate the degradation results. It is critical to look closely at the chemical compositions of the drug substance and drug product.
- Combinations of excipients that will stabilize a formulation and fail to expose a potential stability problem should be avoided. For example, the two lubricants, magnesium stearate and stearic acid, should not be combined if basic excipients are problematic and stearic acid acts as a stabilizer.
- If the amount of material to test is limited or if the final tableting step is rate-limiting, blends can be used to get an initial profile of the stability. Lead and backup formulations can be examined as well to obtain comparison data on formulation stability.
- These results can be communicated back to the formulation group before the nomination of the final clinical or commercial dosage form (depending on the stage of development).
- Therefore, these early drug product degradation studies can have significant time- and cost-saving implications for a pharmaceutical development project.
Global Perspective of Degradation and Impurity Process:-
-
- Figure displays where purposeful degradation studies, method development, and unknown identification fall in the global perspective of analytical compound development.
- Identification of the degradation sample set leads to understanding of degradation mechanisms. Along with process-related impurities/intermediates and knowledge of the drug substance synthesis and drug product formulation, this understanding provides for rational development of chromatographic methods and understanding of the chemical characteristics of the process.
- The sequence of studies and the knowledge gained enable the development of a rationale for regulatory specifications, provide understanding of stability studies, and assist with packaging strategies and creation of impurity grids for filing. Figure is derived from the suggestions put forth in the ICH Draft Consensus Guidelines: Impurities in New Drug Substances and Impurities in New Drug Products[40,41].
Figure no 4 Global perspective of the purposeful degradation, identification, and method development process.
Drug Substance:
Section 3. Rationale for the Reporting and Control of Impurities
- The applicant should summarize those actual and potential impurities most likely to arise during synthesis, purification, and storage of the new drug substance. This summary should be based on sound scientific appraisal of the chemical reactions involved in the synthesis, impurities associated with raw materials which could contribute to the impurity profile of the new drug substance, and possible degradation products.
- This discussion may include only those impurities that may reasonably be expected based on knowledge of the chemical reactions and conditions involved.The applicant should summarize the laboratory studies conducted to detect impurities in the new drug substance. This summary should include test results of batches manufactured during the development process and batches from the commercial process, as well as results of intentional degradation studies used to identify potential impurities arising during storage.
Section 5. Reporting Impurity Content in Batches
- Analytical results should be provided for all batches of the new drug substance used for clinical, safety, and stability testing, as well as for batches representative of the proposed commercial process.
- The content of individual identified and unidentified, and total impurities, observed in these batches of the new drug substance, should be reported with the analytical procedures indicated. A tabulation (e.g., spreadsheet) of the data is recommended.
Drug Product:
Section 2.2. Rationale for the Reporting and Control of Impurities
- The applicant should summarize those degradation products observed during stability studies of the Drug product. This summary should be based on sound scientific appraisal of potential degradation pathways in the Drug product and impurities arising from the interaction with excipients and/or the immediate container/closure system.
- In addition, the applicant should summarize any laboratory studies conducted to detect degradation products in the Drug product. This summary should include test results of batches representative of the proposed commercial process. A rationale should be provided for exclusion of those impurities which are not degradation products, e.g., process impurities from the drug substance and excipients and their related impurities.
- Federal regulations require more from the pharmaceutical industry than just reporting that impurities and degradation products may exist. The 1987 FDA Stability Guideline gives guidance on the procedure to follow when degradation products are detected11:
The following information about them should be submitted when available:
- Identity and chemical structure.
- Cross-reference to any available information about biological effect and significance at the concentrations likely to be encountered.
- Procedure for isolation and purification.
- Mechanism of formation, including order of reaction.
- Physical and chemical properties.
- Specification and directions for testing for their presence at the levels or concentrations expected to be present.
- Indication of pharmacological action or inaction.
CONCLUSION:
In conclusion, the comprehensive overview of degradation investigations for pharmaceutical compounds underscores their critical importance in drug development and quality assurance processes. Through a thorough examination of methodologies, challenges, and recent advancements, it becomes evident that degradation studies serve as a cornerstone in ensuring the stability, safety, and efficacy of drug candidates.
By understanding the various factors influencing drug degradation and employing advanced analytical techniques, researchers can identify degradation pathways, characterize degradation products, and predict stability profiles with greater accuracy. This knowledge enables the optimization of formulations, storage conditions, and manufacturing processes, thereby enhancing the overall quality of pharmaceutical products.
Moreover, adherence to regulatory guidelines and compliance with international standards, such as those provided by the International Council for Harmonisation (ICH), is paramount in conducting degradation studies effectively. Regulatory oversight ensures that pharmaceutical companies meet stringent requirements for product quality, safety, and efficacy.
Looking ahead, continued innovation in analytical methodologies, computational modeling, and data analysis techniques will further enhance the efficiency and reliability of degradation studies. By leveraging these advancements, researchers can address emerging challenges and adapt to evolving regulatory landscapes, ultimately advancing the development and commercialization of safe and effective drug candidates.
In essence, the synthesis of knowledge presented in this overview reaffirms the indispensable role of degradation investigations in pharmaceutical development. Through interdisciplinary collaboration and a commitment to excellence, stakeholders across the industry can navigate the complexities of drug degradation, ultimately delivering high-quality medications that positively impact global health and well-being.
REFERENCES
- International Conference on Harmonization, “Draft Revised Guidance on Impurities in New Drug Substances,” Federal Register 65 (140), 45085-45090 (2000).
- International Conference on Harmonization, “Draft Revised Guidance on Impurities in New Drug Products,” Federal Register 65 (139) 44791-44797 (2000).
- Alsante KM, Hatajik TD, Lohr LL, Sharp TR. Isolation and identification of process related impurities and degradation products from pharmaceutical drug candidates, Part I. American Pharmaceutical Review. 2001;4:70-8.
- International Conference on Harmonisation Steering Committee. Stability Testing of New Drug Substances and Products, 1999.
- USP 24–NF 19. The United States Pharmacopeial Convention, Inc., Rockville, MD, 1999.
- US. Food and Drug Administration. Drug Stability Guidelines, February 1987.
- Knapman, K. Mod. Drug Discovery 3:53–57, 2000.
- Byrn, S., Pfeiffer, R., Ganey, M., Hoiberg, C., and Poochikian, G. Pharm. Res. 12:945–954, 1995.
- Zollinger, H. Color: A Multidisciplinary Approach. Wiley-VCH, New York, 1999.
- Alsante KM, Ando A, Brown R, Ensing J, Hatajik TD, Kong W, et al. The role of degradant profiling in active pharmaceutical ingredients and drug products. Advanced drug delivery reviews. 2007;59(1):29-37.
- Lowry, T. H. and Richardson, K. S. Mechanism and Theory in Organic Chemistry, 3rd ed.Harper & Row, New York, 1987.
- Ahuja, S. Impurities Evaluation of Pharmaceuticals. Marcel Dekker, New York, 1998.
- Allen, J. M. Basic principles of drug photostability testing. Presented at the Photostability 1999 Conference, Washington, DC, 1999.
- International Conference on Harmonisation Steering Committee. Photostability Testing of New Drug Substances and Products, Federal Register 62(95), 27115–27 (1996)
- Allen, J. M. Principles of Actinometry. Photostability 1999 Conference, Washington, DC,1999.
- Wirth, D. D., Baertschi, S.W., Johnson, R. A., Maple, S. R., Miller, M. S., Hallenbeck, D. K., and Gregg, S. M. J. Pharm. Sci. 87: 31–39, 1998.
- Shaw, P. E., Tatum, J. H., and Berry, R. E. Carbohydr. Res. 5:266–273, 1967.
- International Conference on Harmonisation Steering Committee. Draft Revised Guidance on Impurities in New Drug Substances, Federal Register 65(140), 45085–45090 (2000)
- International Conference on Harmonisation Steering Committee. Draft Revised Guidance on Impurities in New Drug Products, Federal Register 65(137), 44791–44797 (2000).


 10.5281/zenodo.10913401
10.5281/zenodo.10913401