
We use cookies to ensure our website works properly and to personalise your experience. Cookies policy
Pharmacology Department, MET’s Institute of D. Pharmacy, Adgaon.
Drug development and regulation are complex, resource-intensive processes that differ greatly between nations. Examining the global drug regulatory environment, this analysis emphasizes the complexities of medication approval, post-market surveillance, and discovery. It looks at how regulatory affairs helps pharmaceutical businesses and regulatory authorities communicate while maintaining adherence to local, national, and international regulations. Important regulatory bodies are covered, along with their approval procedures and regulatory obstacles, such as the FDA (USA), EMA (EU), CDSCO (India), and others. The review highlights the difficulties In approving drugs, including issues with safety, effectiveness, data integrity, and political and economic factors. The report also highlights the increasing significance of gene therapy, personalized medicine, and repurposed medications, illustrating how scientific and technical advancements have affected regulatory procedures. In order to simplify medication laws and improve access to efficient treatments worldwide, it finishes by looking at international harmonization initiatives, especially those carried out by the International Council for Harmonization (ICH). To effectively address the demands of global healthcare, the changing environment of drug regulation necessitates flexible, open, and cooperative approaches.
The traditional drug development process is a complex, expensive, and time-consuming process that involves discovery, computer aided design, advanced screening, and disease research.1 Compliance with regulations for new drug approval varies from country to country creating competitive pressures. Understanding this difference is important for effective drug elimination strategies.2 The pharmaceutical industry is considered one of the most demanding industries in the world. Regulatory bodies ensure that medicines comply with all laws and regulations. Each country has its own regulatory body responsible for issuing guidelines and enforcing laws regarding manufacturing, marketing, registration, licensing, enrollment, drug discovery process and life cycle of drug products.3 Medicines and medicines management specialists are expanding product market service. Repurposed medicines can reduce costs while healthcare providers monitor their efficacy and safety. 30 % of repurposed medicines are approved through randomized controlled trials.4
Regulatory Affairs:
The goal of regulatory affairs is to create a consistent and efficient balance between agency responsiveness to customer demands and voluntary and regulatory compliance and agency responsiveness to consumer requirements. It examines and coordinates all proposed legal proceedings to ensure compliance with regulatory policy.5
The following events may be attended by the regulatory affairs department:
1)Give product development advice.
2)Check the IND application before granting approval for both clinical and non-clinical studies.
3)Provide IND with updates and extensions. Obtain regulatory authority approval.
4)It requires a New Drug Application (NDA) or Marketing Authorization Application (MAA).6
Role of Regulatory Affairs in pharmaceutical industry:
Role of regulatory affairs professional is to act as Cooperation with regulatory agencies:
Regulatory Bodies/Authorities:
Regulatory authority is a regulatory body which conduct the regulatory activities relating to medicines, it includes processing and marketing authorizations, monitoring the adverse drug 4eactions& side effects, inspection, monitoring the safety of medicines and quality testing. It can be created by government to oversee and enforces The regulations regarding occupational health & safety of medicinal products .9
List of international regulatory authorities:
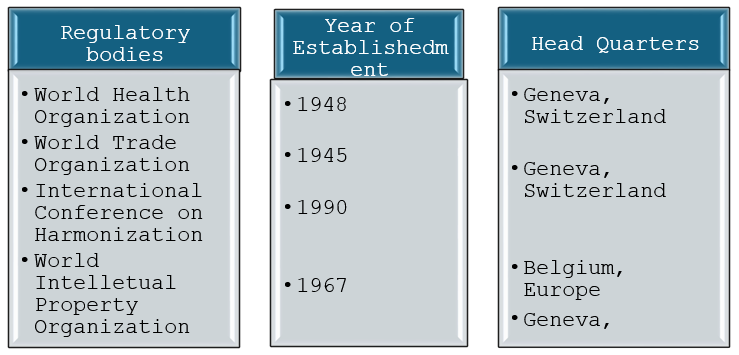
List of Regulatory bodies in other countries:

Drug regulatory bodies have grown more capable of reviewing and approving studies and are now more watchful of globally financed research.
Three separate stages in the development of domestic drug regulators can be distinguished:
1.The first step in the drug regulator’s development was to switch from routine FDA or European Medicines Agency (EMEA) approval of pharmaceuticals or widespread adoption of WHO guidelines to in-country evaluation.
2.Growing political independence of domestic regulators, who lack the resources and know-how to carry out independent reviews but are frequently motivated by worries about possible research misuse and participant misunderstanding
3.The review process’s maturation with growing domestic expertise and capability, frequently aided by exchange initiatives with the FDA, EMEA, or WHO.11
Drug Approval Process:
At the moment, different nations must adhere to various regulatory standards in order for new drugs to be approved. It is nearly impossible to use a unified regulatory methodology for marketing authorization applications (MAAs) across multiple nations. As a result, understanding the legal requirements for MAA in each nation is essential.12
The two developed nations with the strictest drug regulations are the US and the EU.13
Drug Approval in United States:
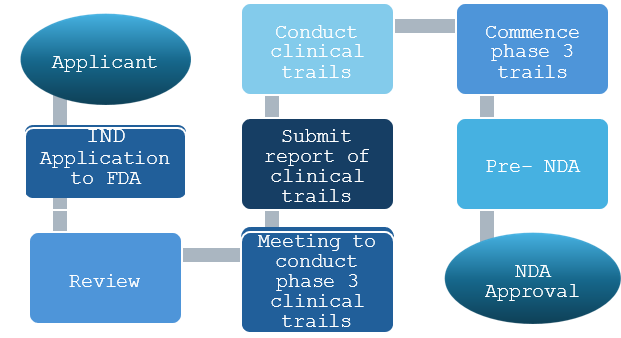
Perhaps the strictest requirements in the world for licensing new medications are found in the United States. Many people believe that the United States has the strictest drug approval requirements in the world. Application of Investigational New Drugs (IND) It is an application submitted to the FDA to begin human clinical trials in the event that preclinical trial results indicate the medicine is safe. The IND application must be submitted by a company or organization known as a Sponsor. It is possible to schedule a pre-IND meeting with the FDA to go over several topics: The methodology of animal research, which is necessary to support clinical investigations Your planned procedure for carrying out the clinical trial The investigational drug’s composition, production, and management a conference like this will assist the sponsor in planning animal studies, collecting information, and creating the clinical protocol in accordance with FDA recommendations. Figure 1 shows a clear flow chart of the IND procedure. Application for New Drugs (NDA) A producer submits a New Drug Application (NDA), which is a formal request to produce and market a new medication in the US, if clinical research indicates that the medication is reasonably safe, effective, and won’t put patients at unreasonable risk. Figure 3 provides an illustration of the NDA process.13,14,15
Abbreviated New Drug Application (ANDA) –
This application is for the approval of generic medications. The original, brand-name product and the clinical trials that were conducted do not have to be replicated by the sponsor. Rather, producers of generic medications are required to verify that their product is identical to and bioequivalent to apreviouslyauthorizedbrand-namemedicine.16

European Union Approval Process:
Clinical trial applications and marketing authorization applications are the two regulatory procedures for drug approval. Applications for marketing permission are authorized at both the member state and centralized levels, whereas applications for clinical trials are approved at the member state level. While Mutual Recognition processes enable applicants to acquire authorization in Concerned Member States (CMS) other than the Reference Member State (RMS), centralized procedures enable applicants to receive a marketing authorization valid throughout the EU. While decentralized procedures enable businesses to concurrently apply for authorization in several EU nations for items that have not yet received authorization in any EU country, nationalized procedures only let applicants to receive marketing authorization in one member state. For final clearance, the EMA opinion must be issued within 210 days.17,18
3)Approval Of New Drug in India:
A company in India must apply for authorization from the licensing authority (DCGI) to manufacture or import a new medicine by submitting Form 44 together with the information provided in Schedule Y of the Drugs and Cosmetics Act 1940 and Rules 1945. It must carry out clinical trials in compliance with the parameters outlined in Schedule Y and submit the results of such clinical trials in the manner required in order to demonstrate its efficacy and safety in the Indian population. However, Rule 122A of the Drugs and Cosmetics Act 1940 and Rules 1945 provide that the licensing authority may waive some of the risks if he determines that allowing the import of novel drugs based on evidence from trials conducted in other nations is in the public’s best interests. Similarly, Rule 122A contains another clause that states that clinical trials may be omitted for newly licensed medications that have been in use for a number of years in other nations.19,20
Regulatory Hurdles: -
1)Approval process: - It is illegal to manufacture or import new medications without the Licensing Authority’s (DCGI) approval. Applicants must submit an application using Form 44, pay the required costs, and submit pertinent information, such as chemical and pharmaceutical data, animal pharmacological and toxicological data, safety and efficacy regulatory status, and local clinical studies, in order to be granted approval.21
A) Challenges In drug approval: -
Preclinical testing, clinical trial design, patient recruitment, data management, regulatory filings, FDA review, safety and efficacy issues, manufacturing and quality control, labeling and packaging, and post-marketing surveillance are some of the challenges that come with the drug approval process. These difficulties can be expensive, time-consuming, and uncertain. Careful planning, patient recruiting, data management, and regulatory considerations must all be addressed for drug development to be successful. Resolving safety and efficacy issues can be difficult, and the FDA review process can be drawn out and unexpected.21
Safety And Efficacy: -
New medicines under U.S. law do not require a specific degree of efficacy, but may be prescribed and marketed with almost no efficacy. This raises suspicion among doctors and patients, as the Federal Food, Drug, and Cosmetic Act states that “substantial evidence that the drug will have the effect it purports” is all that is needed. Open communication and education are crucial for risk management and informed usage. Adverse events, such as complaints, can occur when a product is made available to the public.22,23
Post-Market Surveillance:
Post-market surveillance is crucial for medical gadget manufacturers to ensure safety and prevent potential issues. The FDA has established guidelines for manufacturers to register companies, use tracking systems, and report failures or fatalities. Violations can result in fines, license revocation, or jail time. Post-approval studies are another requirement. Manufacturers often outsource postmarket surveillance to ensure compliance and safety. As most products are sold globally, national regulations are essential for user safety.24,25
Economic And Political Factor-
Political factor: - The FDA’s drug reviews are influenced by political pressures, high uncertainty, and low reversibility. Public pressure can help identify priority drugs, but the agency faces limits on accelerating drug reviews. Delays have political costs and depend on how effectively drug demanders present their case to the agency, Congress, and the media. Prior to the 1980s, FDA delays were rarely criticized, highlighting the agency’s political influence.26
Economic factor/Drug pricing –The pharmaceutical industry is expanding globally, producing new medications and conducting research. However, issues with the current regulatory framework, such as duplicative trials and unnecessary regulation, can lead to increased costs and treatment delays. To protect consumers from harmful medications, countries are attempting to harmonize drug regulations by crossing national boundaries and focusing on a global perspective.27
Global Harmonization –
The international Conference on Harmonization of Technical Requirements for Registration of Pharmaceuticals for Human Use (ICH) was established in 1990 by the FDA, European Communities, and Japanese pharmaceutical producers to address the differences in pharmaceutical values and risk levels among nations. A unified regulatory approach could facilitate quicker access to efficient treatments and improve consumer access. The harmonization process must adopt a global perspective, with representatives from public and scientific sectors participating widely. Developing nations, particularly in Latin America and the Caribbean, can have a say in international issues like pharmaceutical harmonization procedures. Governments must prioritize global healthcare and ensure access to research in developing nations.28,29,30
Technological And Scientific Advancements in Drug Approval
1)Personalized Medicine
Personalized medicine signifies a transformative shift in healthcare, marking a departure from standardized approaches towards individualized treatments. It acknowledges the significance of genetic makeup, lifestyle, and environmental factors in health and treatment response. Utilizing advanced technologies such as genomics and data analytics, personalized medicine aims to innovate healthcare delivery by offering precise and targeted therapies. At its core, personalized medicine relies on a thorough examination of an individual’s genetic composition through genomic sequencing, enabling healthcare professionals to pinpoint specific genetic variations and mutations impacting diseases.31,
Innovations in Personalized Medicine:
2.Gene Therapy
Gene therapy has the potential to transform global healthcare, with over 1,800 active and recruiting trials globally. By 2030, over 60 US approvals are projected, with over 500,000 patients expected to be treated with gene therapies. However, significant scientific and regulatory barriers exist, limiting global market expansion. To overcome these challenges, regulatory frameworks emphasizing predictability, efficiency, and flexibility are needed. Organizations like the American Society of Gene and Cell Therapy (ASGCT) are working to increase dialogue on evolving regulatory frameworks for gene therapies.33
3.Other Innovations
REFERENCES

Dhanashri Patil, Gautami Gholap, Hemant Raut*, MRN Shaikh, Regulatory Challenges in The Approval of New Pharmaceuticals, Int. J. of Pharm. Sci., 2025, Vol 3, Issue 4, 3214-3223 https://doi.org/10.5281/zenodo.15286960
 10.5281/zenodo.15286960
10.5281/zenodo.15286960