
We use cookies to ensure our website works properly and to personalise your experience. Cookies policy
Department of Pharmaceutical Analysis, Centre for Pharmaceutical Sciences, University College of Engineering, Science and Technology (UCESTH), Jawaharlal Nehru Technological University, Hyderabad, Telangana-500085, India.
Forced degradation studies play a critical role in modern pharmaceutical development by providing comprehensive insights into the intrinsic stability of drug substances and drug products. These studies involve the intentional exposure of active pharmaceutical ingredients (APIs) and finished dosage forms to extreme stress conditions such as acidic and alkaline hydrolysis, oxidation, thermal stress, photolysis, and humidity in order to induce controlled degradation. Unlike conventional long-term or accelerated stability studies, forced degradation is designed to rapidly elucidate degradation pathways, mechanisms, and potential degradation products that may arise during manufacturing, storage, or distribution. The information obtained from stress testing is essential for impurity profiling, formulation development, excipient compatibility assessment, packaging selection, and the establishment of appropriate control strategies. Furthermore, forced degradation studies are fundamental to the development and validation of stability-indicating analytical methods, particularly chromatographic techniques such as HPLC and UPLC, ensuring accurate quantification of APIs in the presence of degradation products. Regulatory agencies, guided by International Council for Harmonisation (ICH) recommendations, recognize forced degradation as an integral component of pharmaceutical development and regulatory submissions, although specific experimental conditions remain flexible and science-driven. This review provides a comprehensive overview of the principles governing forced degradation studies, outlines methodological considerations for designing effective stress testing protocols, and discusses analytical perspectives for the detection, separation, and characterization of degradation products using advanced techniques such as LC–MS and NMR. Emphasis is placed on regulatory expectations, analytical challenges, and.ensuring drug quality, safety, and regulatory compliance t emerging trends, highlighting the evolving significance of forced degradation studies in throughout the product lifecycle
A crucial aspect of pharmaceutical development is stability assessment, which has a direct effect on the quality, safety, and efficacy of drug substances and drug products during their shelf life. The unstable nature of chemicals can lead to potency loss, toxic degradation products, and an inability to conform to regulatory standards. The traditional stability experiments, such as long-term and accelerated stability tests, are conducted under anticipated storage conditions to determine the shelf life, but these experiments are not always sufficient to indicate all the possible degradation bases of a drug molecule [1]. To circumvent these restrictions, forced degradation studies (sometimes called stress testing) are carried out in the pre-clinical stages of drug development. Such tests entail subjecting active pharmaceutical ingredients (APIs) and finished dosage forms to extreme environmental factors of acidic and basic hydrolysis, oxidative stress, thermal stress, photolytic exposure, and humidity to cause controlled degradation [2]. Forced degradation is not aimed at replicating the conditions of real-time storage but rather speeding up the chemical degradation to gain an insight into the intrinsic stability of the molecule and what its possible degradation routes are [3]. Forced degradation studies are useful in terms of information on degradation mechanisms and the nature of the degradation products formed under different stress conditions. This knowledge is required in the development of the formulations, the evaluation of excipient compatibility, optimization of the manufacturing process, and the choice of the appropriate packaging system [4]. The behavior of a drug substance under degradation is strongly determined by its chemical structure, physicochemical characteristics, and the existence of labile functional groups, and as such, stress testing is an essential instrument in performing a complete stability profile [5]. The most important use of forced degradation studies is in creating stability-indicating analytical methods (SIMs). A stability-indicating procedure is considered to be an analytical procedure capable of precisely and specifically measuring the active pharmaceutical ingredient in the presence of its degradation products, impurities, and excipients [6]. Forced degradation is essential in the generation of samples that can be used to determine the specificity and selectivity of methods, especially chromatographic methods like high-performance liquid chromatography (HPLC) and ultra-performance liquid chromatography (UPLC) [7]. Regulatively, the international agencies have realized the significance of forced degradation studies as part of an overall stability program. The International Council for Harmonisation (ICH) recommends the use of stress testing to determine the degradation pathways, enhance impurity profiling, and validate analytical procedures, but does not state the conditions of the experiment [8]. As a result, forced degradation experiments are commonly integrated into the pharmaceutical development plan to ascertain product quality and compliance with the regulations [9]. The review aims to broadly contend on the principles of the forced degradation research, summarize the methodological issues to be taken into account in stress testing, and point out analytical views that are taken in the detection and characterization of degradation products. It is also highlighted that the role and relevance of forced degradation studies in contemporary pharmaceutical analysis are changing and regulatory [10].
The regulatory bodies of the world have greatly accepted forced degradation experiments as a key scientific device in the pharmaceutical development field to comprehend the degradation pattern and verify the dependability of the stability-reporting analytical techniques. Even though forced degradation is not meant to determine the shelf life, regulatory authorities have made it clear that forced degradation helps in the determination of the degradation pathways, impurity profiling, and specificity of the analytical methods used in the development of a drug and regulatory submission [11].
ICH is the main source of regulation of stability testing and stress studies. ICH Q1A(R2) suggests stress testing of drug substances to determine intrinsic stability properties and determine the possible products of degradation, although exact experimental conditions are not spelt out [12]. It is believed to be important to stress test under hydrolytic, oxidative, thermal, and photolytic conditions to get useful information about degradation. Photostability test requirements are outlined in the ICH Q1B that requires the test of light sensitivity of drug substances and products to identify the necessity of the light-protective packaging or labeling [13]. Besides, ICH Q2(R2) emphasizes specificity in validation of analytical methods, which is usually illustrated with the help of the samples produced as a result of forced degradation studies [14].
Regulatory bodies, including the United States Food and Drug Administration (US FDA) and European Medicines Agency (EMA), require forced degradation testing to be included in the pharmaceutical development programs, especially in new drug applications (NDAs) and abbreviated new drug applications (ANDAs). These works assist impurity justification, degradation pathway explanation, and validation of stability-indicating techniques employed in quality control [15].
The current regulatory trends lead to a risk-based and science-based approach to forced degradation and prompt manufacturers to design stress conditions unique to each chemical nature of the active pharmaceutical ingredient (API) and dosage form, instead of using the same stress conditions on all products [16]. This methodology is well in line with the Quality by Design (QbD) strategies, in which a systematic knowledge of the degradation behavior refines the method's strength and regulatory standards.
The importance of forced degradation studies to impurity profiling and adherence to impurity-directed regulatory guidelines is great. The regulatory guidelines emphasize the need to determine, define, and manage the degradation products, which could be created during the storage or processing process [17]. The data of stress testing is often applied to define the levels of degradation products and support the impurity limits in regulatory applications. The development of analytical methods has also contributed to expectations on regulatory standards, as the agencies are now more willing to support the application of hyphenated methods, including LCMS and high-resolution mass spectrometry (to characterize degradation products in case of substantial degradation) [18]. All these expectations mirror the increasing trend of scientific justification and a thorough evaluation of impurity in pharmaceutical analysis.
In regulatory dosses, forced degradation data are often included to assist in analytical methods validation and control of impurities. Stress conditions, degradation level, and methodology of analysis should be well documented, and the results should be interpreted to prevent regulatory questions or postponement of approval [19]. The development of forced degradation studies contributes to regulatory confidence by showing comprehensive knowledge of product stability in the drug lifecycle. As a whole, regulatory considerations and recent academic findings support the essence of the forced degradation studies as one of the essential elements of the pharmaceutical development, which combines stability testing with the validation of the analytical methods together with regulatory compliance [20].
Forced degradation (stress testing) is conducted to deliberately subject drug substances or drug products to degradation conditions in controlled conditions to obtain an intrinsic profile of their stability. Stress testing is not several weeks of stability testing as with routine stability tests, but rather aimed at producing the degradation products in a short period, or by exposing the material to harsh conditions. The principal idea is the exposure of the drug to chemical and physical stress factors that simulate the possible degradation routes, enabling the determination of the potential degradation tendencies during the manufacturing, storage, and processing [21].
The key objectives of forced degradation studies include:
The significant pathways of degradation that are usually noticed during forced degradation studies are:
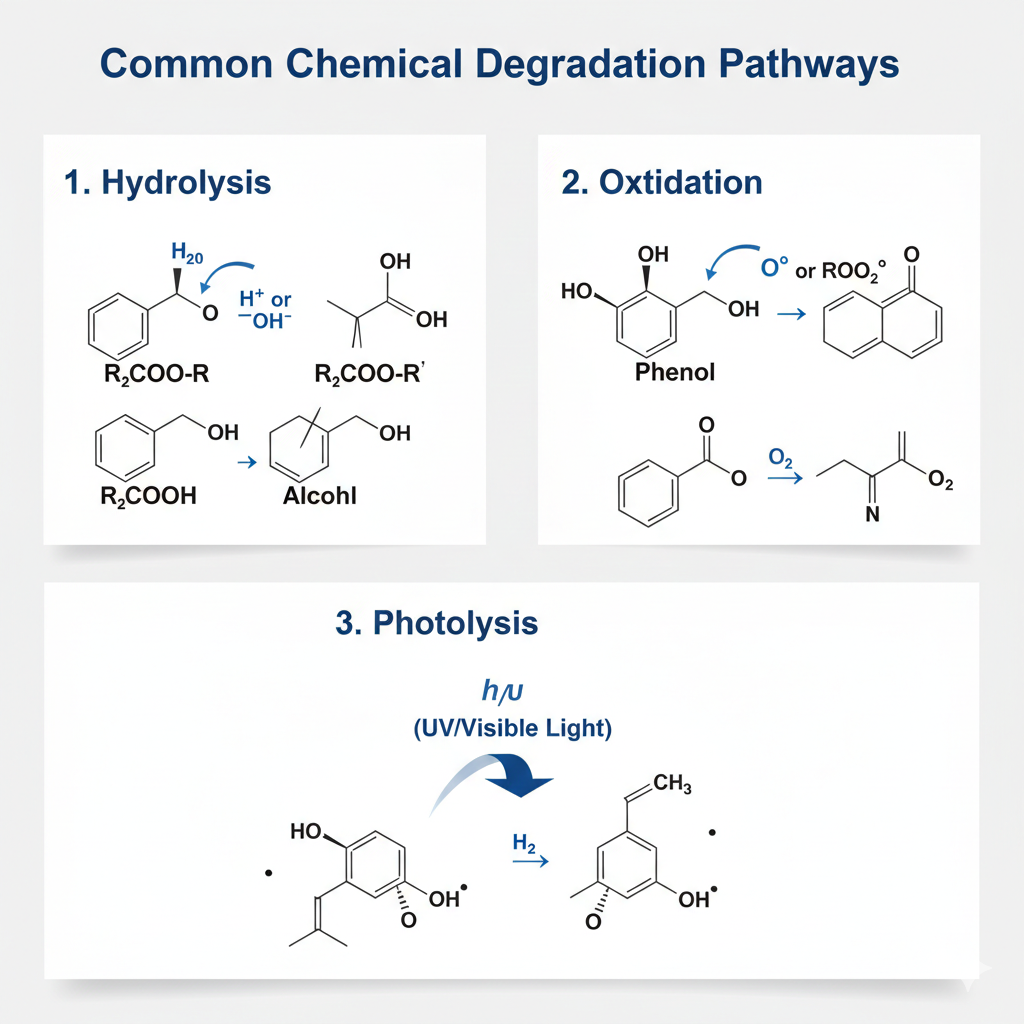
Figure 1: Major chemical degradation pathways including hydrolytic cleavage and oxidative radical mechanisms.
The conditions of forced degradation must be selected with caution in order to obtain moderate degradation (usually 101030 percent), enough to test analytical methods but not to yield secondary products of excessive degradation. The stress conditions shall be optimised based on the chemical nature and dosage form of the drug. Over-stressing can cause the formation of irrelevant degradation products that do not accurately depict the stability behavior [31].
Knowledge of degradation kinetics aids in forecasting stability under normal storage conditions and in determining factors that are critical and enhance degradation. The mechanistic understanding can also be useful in creating strong formulations and protective approaches, e.g., antioxidants, pH modifications, and protective packaging [32].
Forced degradation studies are supposed to be designed depending on the dosage form. As an example, liquid formulations will be susceptible to hydrolysis and oxidation, whereas solid dosage forms can be subject to photolytic or moisture degradation. Biopharmaceuticals are commonly subjected to special stress conditions in order to measure protein denaturation, aggregation, or chemical modifications [33].
Forced degradation studies are carried out to purposely cause active pharmaceutical ingredients (APIs) and finished dosage forms to degrade under any of the controlled stress conditions. These studies aim at producing the degradation products in a short time and give information on degradation pathways, which is utilized in the establishment of stability-indicating methods and formulation optimization. Forced degradation methodology entails ensuring that the right stress conditions are selected, the sample is prepared, the parameters of stress are optimized, and the evaluation criteria are considered to ensure that meaningful degradation is achieved, but without over-stressing the molecule [34].
The choice of the stress conditions is based on the chemical structure of the API, degradation vulnerabilities that are known, and the dosage form. Stress conditions are usually chosen to potentially simulate probable degradation pathways, including hydrolysis, oxidation, photolysis, thermal degradation, and humidity-influenced degradation [35]. Examples of common stress conditions are:
Table 1: Forced Degradation Stress Conditions
|
Stress Type |
Common Reagents / Conditions |
Recommended Endpoint |
Source (Ref. No.) |
|
Acid Hydrolysis |
0.1–1 N HCl; room or elevated temperature |
10–30% Degradation |
[8, 36, 46] |
|
Base Hydrolysis |
0.1–1 N NaOH or KOH; room or elevated temperature |
10–30% Degradation |
[12, 36, 46] |
|
Oxidative Stress |
0.1–3% $H_2O_2$ or metal-catalyzed systems |
10–30% Degradation |
[37, 46] |
|
Photostability |
UV and Visible light as per ICH Q1B |
Moderate Degradation |
[13, 38] |
|
Thermal Stress |
40–80°C; dry heat or controlled humidity |
10–30% Degradation |
[29, 39, 46] |
|
Humidity |
High RH (e.g., 75%) at elevated temperatures |
Moderate Degradation |
[30, 40] |
Sample preparation is very important to achieve credible and repeatable forced degradation results. The preparation of the samples will be based on the type of research conducted on the API or the final dosage form.
Using API testing, intrinsic stability is displayed without interference by excipients, and dosage form testing offers the actual behavior of stability in the real world with the influence of excipients [41].
The solvents employed in forced degradation should also be non-reactive, and the solvents should not react with the API. The common solvents are water, methanol, acetonitrile, or buffer solutions based on the solubility and stability of the drug. During the hydrolysis studies, the pH needs to be regulated [42].
Once stress conditions have been applied, the degradation process must be terminated by applying neutralization or quenching. In the case of acid/base hydrolysis, neutralization is done with a base/acid of equal value. In oxidative stress reaction, the reaction is terminated by the use of quenching agents like sodium bisulfite or catalase. Quenching stops additional degradation in the process of analysis and sample processing [43].
Optimization aims at producing a moderate degradation (10-30%) that is enough to create a method, but not too much to create a lot of secondary degradation. Optimization involves modification of time, temperature, and concentration of stress agents.
The temperature and duration of stress are manipulated to have the required degree of degradation. Increased degradation is caused by high temperature and the length of time of exposure, and conditions should be maintained to prevent excessive degradation [44].
Controlled degradation is undertaken by the concentration of acid, base, oxidizing agents, or buffer strength. Slow change in concentration can be used to recognize the best stress conditions [45].
The degradation in the range of 1030% is considered optimal to develop stability-indicating methods because it creates a sufficient amount of degradation products to separate and identify, but enough API to be detected [46].
In forced degradation experiments, intricate mixtures of degradation products are produced and hence require sophisticated analytical instruments to separate, detect, and characterize them. Along with the development of stability-indicating methods (SIM), analytical techniques are also used to assist in regulatory compliance due to evidence of specificity, selectivity, and impurity profiling [48].
A stability-indicating method is a tested analytical process and can properly measure the API with its degradation products, impurities, and excipients. Forced degradation experiments furnish the necessary degraded samples to show the specificity and selectivity of the method to make the correct quantification in stability experiments [49].

Figure 2: Systematic workflow for developing Stability-Indicating Methods (SIMs) using forced degradation samples.
In the cases where the chromatographic separation may not be enough to identify degradation products, other sophisticated methods of structural elucidation and confirmation are used: LC-MS/MS, HRMS, and NMR [51].
Table 2: Comparison of Analytical Techniques for Degradation Profiling
|
Technique |
Primary Application |
Key Advantage |
Source (Ref. No.) |
|
HPLC / UPLC |
Routine SIM development and quantification. |
High resolution and the ability to separate multiple products. |
[7, 50] |
|
LC-MS/MS / HRMS |
Identification of unknown degradation products. |
Provides accurate mass and fragmentation data. |
[18, 52] |
|
NMR Spectroscopy |
Confirmatory structural elucidation. |
Differentiates isomeric or structurally similar degradants. |
[53, 57] |
|
Hyphenated Tools |
Integrated separation and structural analysis. |
Essential for characterization of complex mixtures. |
[55] |
These degradation products should be identified using a combination of chromatographic and spectroscopic methods. Chromatography gives the information on retention time and purity, and spectroscopic techniques verify the structure of the molecule and its decomposition. The integrated method makes sure that all degradation pathways are identified correctly [54].
LC-MS, GC-MS, and LC-NMR are hyphenated methods that combine separation and structural analysis, making it possible to easily identify complex degradation products. These methods are critical to precise characterization and substance regulation expectations of impurity profiling and records of stability [55].
To perform a safety assessment and validate the method, in-depth impurity profiling and identification of degradation products are needed by regulatory agencies. Impurity limits, stability claims, and quality control strategies in the regulatory submissions are supported by analytical data of forced degradation studies [56].
The product mixtures formed in forced degradation processes tend to be complicated and degraded with low levels of degradation products, which are difficult to detect and identify. These problems are countered using high sensitivity, structural confirmation, and precise mass data of unknown degradants using advanced techniques such as HRMS and NMR [57].
In pharmaceutical research, forced degradation testing is commonly applied to measure the stability of drug substances and products in stressful conditions. Such studies are useful in the study of chemical behavior of drugs, the anticipation of potential degradation routes, and the development of mechanisms to enhance stability during formulation, manufacturing, packaging, and storage [58].
During formulation development, forced degradation assists in screening excipients and the choice of stabilizers to inhibit degradation. The API, excipients, and additives compatibility is assessed based on the stress conditions of the formulation, and utilizing the degradation profile [59]. This allows formulation composition to be optimized in order to improve stability and shelf life.
Forced degradation studies are used to determine process-related degradation in the manufacturing process, e.g., heat exposure during drying, compressional force during tablet production, and moisture exposure. Knowledge of process-induced degradation assists in setting up control measures and critical process parameters (CPPs) to reduce degradation in the manufacturing process [60].
Forced degradation researches are used to create adequate packaging systems as they consider sensitivity to light, humidity, and oxygen. In case the drug is found to be degraded by sunlight or moisture, it is possible to select protective packaging, such as amber glass, foil laminate, or desiccants, to maintain stability throughout storage and distribution [61].
Forced degradation tests are used to help profile impurities, in that they produce degradation products that can occur during storage or processing. These are determined and measured to assess the toxicological hazard of the products and to comply with regulatory levels of impurities. This plays a role in quality control by safety assessment and risk-based quality control [62].
The results of forced degradation experiments aid in developing a powerful control program regarding stability and quality. The control strategies are the establishment of storage conditions, packaging, shelf life definition, and specifying the degradation product limits in the drug product [63].
The forced degradation studies make it possible to have a risk-based approach through establishing the key degradation pathway and triggers, which can be implemented by formulation scientists to prevent them. This is in line with the Quality by Design (QbD) philosophy, whereby it is guaranteed that the product will be stable in a real-life situation [64].
Forced degradation data are valuable during the product life cycle, such as during the post-approval modifications that include new formulation, new manufacturing location, or revised packaging. Such researches present scientific rationale for change and regulatory submissions [65].
The forced degradation studies offer empirical information on the degradation behavior, development of analytical methods, and the degradation products of various classes of pharmaceuticals. The choice of case studies illustrates the application of stress testing in determining degradation pathways and helping to formulate and make analytical decisions.
Doxorubicin is an anti-cancer drug that is an anthracycline, and studies have been performed to assess its stability under various stress conditions throughout forced degradation. As part of a comprehensive forced degradation test, doxorubicin was exposed to acidic, alkaline, neutral hydrolytic, oxidative, thermal, and photolytic conditions as per ICH Q1A(R2) to determine the degradation pathways and products [66]. The experiment showed that doxorubicin was very susceptible to alkaline hydrolysis and oxidative stress and generated several degradation products. Liquid chromatography was used to separate four products of the oxidative degradation and one product of the hydrolytic degradation, and the products were identified by use of mass spectrometry, which revealed the usefulness of forced degradation in elucidating the structures of degradants [66]. The case points out that forced degradation will be used to detect possible impurities that may not be evident in the context of normal storage but are essential in the validation of methods and assessment of safety.
A recent review of the literature on the forced degradation of anti-cancer drugs demonstrates typical forces of degradation and their assessment based on analytical technologies. The complexity of structures and the presence of labile functional groups render such anti-cancer molecules as doxorubicin and paclitaxel susceptible to hydrolytic, oxidative, and photolytic degradation pathways. Forced stress conditions supply information about the stability and product degradation products that are of interest to the formulation design and regulatory compliance [67]. Analytical techniques such as HPLC and LCMS were also reported in the review, which are usually used to separate and label the degradation products of these drugs in controlled stress conditions [67]. This case highlights the number of degradation pathways that complex therapeutic agents may undergo and the type of analytical technique that is needed to solve the problem.
The amlodipine case exemplifies forced degradation studies of the antihypertensive drug amlodipine, where various stress conditions can be used to demonstrate different degradation pathways. Amlodipine in acidic, alkaline, neutral hydrolytic, oxidative, and photolytic conditions produced various degradation products, some of which were alkaline and oxidative degradants. These compounds were characterized and monitored with the help of LCMS and hyphenated methods, which showed that stress studies can be used to identify pathway-specific degradation products as a means of structural elucidation and method validation [68]. In spite of the fact that this is a literature example regarding the application of the method, it shows the significance of a wide range of stress conditions to obtain full periability profiles of cardiovascular drugs.
Other degradants not always found in aqueous conditions may be found in some forced degradation studies in which the absence of water results in an anhydrous stress environment. In forced degradation of an experimental drug called C1 in an anhydrous state, both acidic and oxidative degradants akin to those observed under the aqueous environment were observed. Nevertheless, anhydrous reactions produced more degradation products, which were not stable in aqueous solutions but could be observed in RP HPLC and mass spectrometry [69]. This case shows that alternative stress environments may provide distinctive information about the entire profile of degradation and must be taken into account when developing comprehensive stress testing approaches.
The effects of individual excipients and formulation variables on stability can be determined through mechanistic forced degradation studies, e.g., those investigating oxidative degradation pathways. As an example, oxidative degradation of an amorphous drug that was spray-dried in a polymer matrix demonstrated that there were numerous oxidative degradants. Researchers were able to use a forced degradation test to find three oxidative degradation products and to test antioxidants like propyl gallate and EDTA to suppress degradation. Of particular focus in the study is the extension of forced degradation to formulation optimization in terms of assessing how stabilizers influence degradation pathways [70]. The case illustrates how forced degradation coupled with formulation design can be used to improve the stability of the product.
The forced degradation studies are very crucial in the development of pharmaceuticals, although they also have some scientific, analytical, and practical issues, which should be well taken care of to achieve significant results.
The main drawback with forced degradation is the definition of the level of stress to be imposed. The selection of too high stress may result in the formation of degradation products that are not relevant pharmaceutically and may not be formed in actual conditions of storage and use. It can result in poor understanding of the degradation mechanism and unrealistic stability techniques [72].
The development of forced degradation experiments to produce scientifically valid and interpretable data is complicated, especially in situations where there is an interaction between several stressors or the association between structure and function is subtle. One-factor methods at a time might not be sufficient to represent interaction, and a more sophisticated design of experiments (DoE) is becoming needed to explain multifactor effects [71].
The guidelines of the regulation, like the ICH Q1A(R2), are general expectations of stress testing without any prescription of stress conditions or performance standards. Such absence of detailed protocol instructions might lead to the inconsistent execution and interpretation of the data among the labs, with an impact on the reproducibility and acceptance by the regulatory authorities [73].
Forced degradation tends to give more intricate blends of products in low concentrations of degradation that makes them difficult to separate and locate by analytical approaches. The identification and characterisation of such products is a highly sensitive task that needs extremely sensitive analysis methodologies (e.g., LCMS, HRMS), and the generation of robust analytical tools that can reflect all the degradants in question is resource-demanding [74].
The interpretation of degradation results to be submitted by the regulators is a scientific interpretation regarding the relevance of the products that were formed in the conditions of stress. It can be hard to differentiate between degradants that are likely to occur under real storage conditions and artefacts of forced conditions, which necessitate proper documentation and justification to prevent any questions that may arise on the part of the regulatory authorities [75].
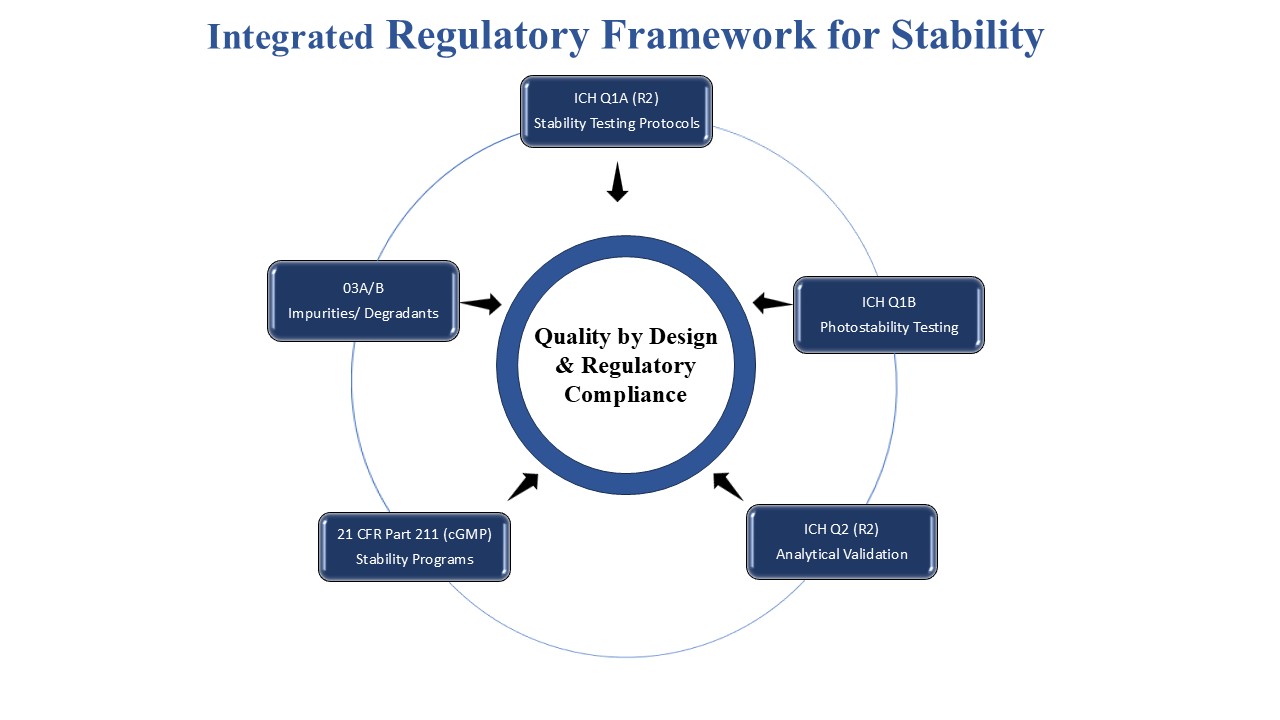
Figure 3: The integration of various ICH guidelines in pharmaceutical stability assessment.
Forced degradation studies are increasingly benefiting from artificial intelligence and machine learning, where AI driven predictive analytics can analyze physicochemical, formulation, and environmental data to forecast degradation behavior, support accelerated stability assessment, and optimize stress testing design [76] these predictive models have also been applied in broader pharmaceutical stability studies to estimate shelf life, guide real time stability monitoring, and identify formulation attributes that influence degradation outcomes [76] in addition, in silico prediction tools that use rule-based degradation knowledge bases such as Zeneth demonstrate how computational methods can generate theoretical degradation products and inform experimental strategy, although continuous refinement and validation are needed to improve prediction accuracy [77] further integration of real time monitoring and predictive analytics allows continuous updating of stability predictions during manufacturing and storage, enhancing quality assurance and enabling adaptive control strategies [78] as these data driven approaches mature, regulatory science and guidance frameworks are expected to evolve to accommodate AI assisted prediction and modeling aligned with Quality by Design principles, encouraging broader adoption of predictive degradation risk assessment in regulatory submissions [79] predictive modeling applied beyond forced degradation such as computational stability forecasting in product development and packaging optimization illustrates how data analytics can improve formulation decision making and stability outcomes and systematic reviews of ML and AI applications highlight the potential and gaps in adopting predictive technologies across pharmaceutical functions, underscoring the importance of robust models, validation, and regulatory adaptation to fully leverage predictive analytics for stability and degradation profiling [80].

Figure 4: Workflow for integrating AI and machine learning into stability and degradation profiling.
CONCLUSION
The forced degradation study is important in modern pharmaceutical development through the systematic uncovering of degradation pathways, the detection of potential impurities, and the creation of stability-indicating analytical techniques necessary in quality control. The studies are part and parcel to formulation design, excipient compatibility testing, manufacturing process, and packaging selection since they furnish essential evidence in ensuring the safety and efficacy of drugs throughout the product lifecycle [81]. The development of the analysis methods, in particular, hyphenated approaches to degradation product identification and mechanistic insight, has helped to enhance the efficiency of the identification of degradation products and comprehension of the mechanism of action greatly, allowing to characterize and validation of the methods in a rigorous manner [85]. The above improvements do not eliminate the fact that over-stress conditions, irrelevant degradation products production, and non-standardized stress protocols remain present and restrict the predictability and comparability of forced degradation results across various laboratories [84]. Here, a future development in which AI-based predictive models and in-silico degradation tools are integrated can be seen as a promising path forward, as it would allow early prediction of degradation pathways, help reduce the experimental load, and allow data-driven decision making throughout drug development [83]. Thus, forced degradation research cannot be eliminated in terms of assuring the quality of pharmaceuticals, and further research, technological advancement, and harmonization of regulatory methods are necessary to further the use of forced degradation research in drug stability testing and regulatory compliance [82].
REFERENCES


Supriya Nidadavolu*, M. Sunitha Reddy, Review on Forced Degradation Studies: Principles, Methodology, and Analytical Perspectives, Int. J. of Pharm. Sci., 2026, Vol 4, Issue 2, 91-110. https://doi.org/10.5281/zenodo.18453996
 10.5281/zenodo.18453996
10.5281/zenodo.18453996