
We use cookies to ensure our website works properly and to personalise your experience. Cookies policy
Modern College of Pharmacy, Pune, Maharashtra.
Solubility and dissolution characteristics of any drug are critical factors controlling its oral bioavailability. Disposition rate of poorly soluble, highly permeable (BCS-II) drugs like atorvastatin calcium can be enhanced by the use of the liquisolid (LS) method. Various liquisolid compacts were designed utilizing a mathematical model for estimation of necessary amounts of powder and liquid ingredients to yield an acceptably low and compressible mixture. Avicel PH 102, Aerosil 200 and Explotab were utilized as carrier, coating material and disintegrant, respectively. Prepared liquisolid systems were characterized for their micromeritic properties and potential drug-excipient interaction using Infrared spectra (IR) analysis, differential scanning calorimetry (DSC) and X- ray powder diffraction (XRPD). Liquisolid compacts were formulated and characterized for their tabletting property. The liquisolid system had good micromeritic properties. IR and DSC analyses eliminated any signiicant drug-excipient interaction. The XRPD analysis confirmed the formation of a solid solution within the compact matrix. The tabletting behavior of the liquisolid compacts was found to be within the acceptable range. Release rates of liquisolid compacts were significantly greater in comparison with directly compressed tablets, owing to improving wetting characteristics and drug surface area. From the derived pharmacokinetic parameters, like the AUC, tmax and Cmax, liquisolid compacts exhibited improved bioavailability than the conventional formulation. This research indicates that the liquisolid method is a good alternative for the enhancement of the dissolution and oral bioavailability of water insoluble drugs a confirmed by the estimation of the pharmacokinetic parameters in vivo in rabbits.
The poor dissolution properties of water in soluble medicines are one of the biggest challenges facing pharmaceutical science. Powder solutions are formulated to hold liquid medicine in powder form and there have been numerous developments for enhancing drug dissolution rate profiles. Over the last few years several methods have been developed including drug micronization, solid dispersion, co-precipitation, lyophilization, micro–encapsulation, application of pro-drug and drug derivatization processes, and inclusion of drug solutions to soft gelatin capsules. Micronization is the most frequent technique employed to enhance the drug's surface area, but this is less efficient when they are designed as tablets or encapsulations (Aguiar et al., 1979; Finholt, 1968; Lin et al., 1968). With respect to soft gelatin capsules, Ebert's (1977) review mentioned that these products exhibited the most effective bio availability as the drug is already in solution form. The soft gelatin capsules, however, are costly to make and need advanced technology. As it is, however, there are best practices to make the liquid oily drugs and drug solutions of water insoluble solid drugs.
Bio-Pharmaceutical classification class II (water insoluble drugs), rate of oral absorption is usually regulated by rate in the GIT. Thus, coupled with permeation, solubility profile and dissolution rate profiles of drugs are significant key determinants of its oral bio-availability. powdered solutions allow to transform drug solutions or liquid dosage forms into moderately flowing powders through an admixture with certain powder excipient. A similar method has been employed by some investigators for enhancing release profiles of a number of water–insoluble drugs (Spireas, 2002).
One of the recent modern methods, Licensed "Powder Solution Technology" has been adopted to prepare water-insoluble drugs in the form of immediate release solid dosage forms. The Liquisolid methods are regarded as smoothly flowing and compressible powders of liquid medications. Such liquid medications can be transformed back into dry – appearing or moistureless, non-adherent free flowing and easily compressible powders by a mere admixture with specifically chosen carriers and coating materials.
In this method, the drug could be in solid dosage form dissolved in solution or a solvent. Drug is solubilized in a maximum molecularly dispersed state. Thus, this is because of their considerably enhanced wetting properties and enhanced surface area of drug available for dissolution. Water– insoluble or poorly water soluble drugs can be anticipated to have enhanced dissolution rate properties as well as enhanced bioavailability.
The Liqui-solid compacts contain two significant formulation components i.e., powder substrate and liquid medication. The powder substrate generally contains: a) compression enhancing, large preferably porous carrier particles; b) flow enhancing, very fine, highly adsorptive coating material particles. Liqui-solid preparations can be hindered by their erratic and poor flow and compaction properties. Inherent to the finished powder / liquid admixtures are fixed.
The weights of different excipients used in powder solution formulations are calculated according to a new mathematical model expression (Liao, 1983).
The molecules of the absorbate diffuse within the absorbent and are finally trapped by the powder particles in their bulk, and hence absorption of the liquid takes place. Adsorption is the process in which liquid is not actually absorbed and instead of being spread throughout the interior of the solid, the molecules only attach to its surface, internal and external. But based on sorbent properties, both of these operations can take place simultaneously, and is called sorption. First the liquid is absorbed in the interior of the particles trapped by its internal structure and upon saturation of this operation adsorption of the liquid onto the external and internal surfaces of the porous carrier particles (Rania et al., 2008)
2. MATERIALS AND METHOD :-
2.1 Materials :
The following samples were received Atovastatin calcium, Polyethylene glycol 400 (PEG400), Methanol, Acetonitrile, Lactose, Silica gel, Sodium starch glycolate, Sodium alginate, Croscarmellose sodium, Crospovidone, Microcrystalline cellulose, Magnesium stearate, Talc powder. All reagents and used were of analytical grade.
2.2 Methods :-
2.2.1: Phase Solubility studies
Solubility studies of Atorvastatin calcium were conducted in distilled water, methanol, phosphate buffer at pH 6.2, and phosphate buffer at pH 7.4. Saturated solutions prepared in these vehicles were stored in a volumetric flask for 48 hours at room temperature (25°C). The solutions were filtered, and their concentration was measured using a UV spectrophotometer at 239 nm. The results were extrapolated to find the present w/w of Atorvastatin calcium in its saturated solution with the solvent being tested.
2.2.2: Preparation of liquisolid based compact
Suitable drug candidate is dispersed in suitable non-volatile solvent like polysorbate 80, PEG 200, etc. having different drug (API) : solvent ratio.
In this step, suitable carrier material with other excipients is added into initial mixture of drug and non-volatile solvent. During this continuous mixing in mortar should br going on.
In this third step, suitable super disintegrants like sodium starch glycolate or micro-crystalline cellulose is added in the prepared mixture with continuous shaking in a mortar.
In this step, suitable coating material is added which absorbs the layer of excess non-volatile solvent over the carrier material. Due to this ,the liquid layer gets converted in the solid layer gets converted in the solid layer and this gives the dry, non-adherent, free flowing powder particles.
The final mixture is then allowed to compress by using tablet compression machine. The prepared liquisolid tablet is then evaluated in graduated for its solubility, dissolution, compressibility.
2.2.3: Pre-compression studies
The flow properties of the liquisolid systems - The flow properties of the liquisolid systems were estimated by determining the angle of repose, Carrs’s index and Hausner’s ratio . The angle of repose was measured by the fixed funnel and freestandind cone method . The Bulk density and Tap densities were determined for the calculation of Hausner’s ration and Carr’s index.
2.2.4: UV spectroscopy
Analytical method validation (UV):- The validation of analytical method was done as per ICH guideline. All the validation parameters such as accuracy, precision, linearity, range, limit of detection and limit of quantitation were determined.
Preparation of Standard Stock Solution:
Standard stock solution containing 100μg/ml of Atovastatin calcium was prepared by dissolving 2.5mg of drug in minimum amount of methanol. Volume was made up to 25 ml with distilled water. Different aliquots were taken from the stock solution and diluted to 10ml with distilled water to prepare a series of dilutions (2µg/ml- 60µg/ml). The absorbances of the samples were recorded at λmax 239 nm.
2.2.5: Infra red spectra analysis:
The infra-red spectra of the solid dispersions were scanned by KBr method employing a Fourier transform infrared spectrophotometer. Base-line correction was done with dried potassium bromide and then the spectrum of pure ATR, liquisolid system was recorded.
2.2.6:- X-ray powder diffraction:
X-ray diffractograms of ATR , and liquisolid formulation were obtained using analytical XRD instrument. The scanning range was from 5-80 degrees.
2.2.7: Differential scanning calorimetry (DSC):
Thermo grams of the samples (ATR and liquisolid compact) were measured on a DSC .The thermal behaviour of the samples were examined at a scanning rate of 100C/min, encompassing a temperature of 0-3000C.
2.2.8: In vitro evaluation of liquisolid compacts:
1) Friability and hardness:
The hardness of prepared liquisolid tablets was measured with a Pizer hardness tester, and the average hardness of three tablets was calculated. The friability of the liquisolid tablets so prepared was determined in a Roche type apparatus and the weight loss percentage was calculated and employed as a measure of friability and the results for all the batches of ATR liquisolid compacts.
2) Weight variation:
Weight variation test was performed according to USP and results of all the batches of ATR liquisolid compacts.
3) Disintegration test:
Disintegration test was performed with disintegration test apparatus as speciied in Indian Pharmacopoeia and results of all the batches of ATR liquisolid compacts.
4) Phase solubility studies:
Phase solubility studies were performed by the method previously reported by Higuchi and Connors (1965). Briefly, excess amounts of Atovasttain calcium were added to 20 ml of solutions i;e distilled water, phosphate buffer pH 7.4, pH 5 & 0.1 N HCl containing various concentrations . The drug to excipient ratios were 1:1, 1:2, 1:4, 1:6, 1:12, 1:18. The suspensions were vigorously shaken for 48 hrs on rotary shaker. After equilibrium was attained, the samples were filtered through a whattman filtered paper (#41). Racecadotril concentration was determined spectrophotometrically at 239nm using UV/VIS double beam Spectro-photometer. After obtaining the results phase solubility constants in different solvent systems and phase solubility relationship were studied.
5) Assay or drug content:
Accurately weighed complexes equivalent to 2.5 mg of Atovastatin calcium were transferred in 25 ml volumetric flask. 10 ml methanol was added and sonicated for 5 min. Volume was made up to 25ml with distilled water. This solution was filtered using whattmen filtere paper in 25 ml volumetric flak, again make up to the volume with distilled water up to 25ml mark. 1ml of solution was withdraw and diluted up to 10 ml with distilled water (10µg/ml) and determined spectrophotometrically at 239 nm. (Same procedure was applied for all the formed complexes).
6) Dissolution study:
The in-vitro release profiles of ATR from liquisolid compacts and direct compression tablets were determined by a dissolution test apparatus USP-II . The study on dissolution was conducted in 900 ml, 450ml and 300ml phosphate buffer of pH 6.2 and distilled water as the dissolution fluid at 37?C ± 2?C and 50 r/min. Then5 ml samples were withdrawn for a period of 60 min at 2-min intervals until 30 min and 15 min intervals between 30 and 60 min. The dissolution medium was replenished with 5 ml of fresh dissolution liquid to ensure sink conditions. The withdrawn samples were filtered and spectrophotometrically (Jasco v430, Japan) analyzed at 239 nm.The average of three determinations was employed to calculatethe release of drug from each of the formulations.
RESULT AND CONCLUSION:-
1. Powder characterization:
The result of powder characterization of Atovaatatin calcium trihydrate shown in Table below.
|
Sr. No |
Powder characterization |
Observation/ Inferences |
|
1 |
Melting point |
155.80C |
|
2 |
pH |
7.12 |
|
3 |
Colour |
White |
|
4 |
Odour |
Odorless |
|
5 |
Texture |
Crystalline |
|
6 |
Bulk density |
0.82 gm |
|
7 |
Tap density |
1.086 gm |
|
8 |
Carr’s index |
24.4 |
|
9 |
Hausner ratio |
1.32 |
|
10 |
Angle of repose |
41.3 |
From the powder characterization data, it was observed that drug was having very poor flow properties.
2. Analytical method validation (UV):
|
Sr. No |
Concentration |
Absorbance at 239 nm |
|||
|
|
|
Water |
Methanol |
Phosphate buffer pH 6.2 |
Phosphate buffer pH 7.4 |
|
1 |
0.2 |
0.0046 |
0.0288 |
0.1737 |
0.0003 |
|
2 |
0.4 |
0.350 |
0.1748 |
0.1912 |
0.2332 |
|
3 |
0.6 |
0.0470 |
0.1822 |
0.3015 |
0.4498 |
|
4 |
0.8 |
0.0335 |
0.1932 |
0.4894 |
0.7064 |
|
5 |
1.0 |
0.1127 |
0.2125 |
0.7356 |
0.9450 |
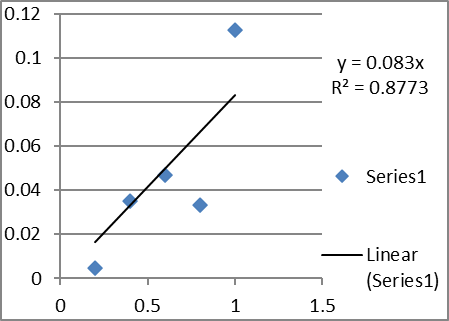
A. Calibration curve in water at λmax 239nm

B. Calibration curve in water a t λmax 239nm
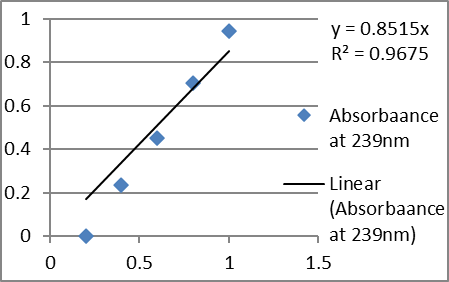
C. Calibration curve in phosphate buffer pH 6.2 at λmax 239nm
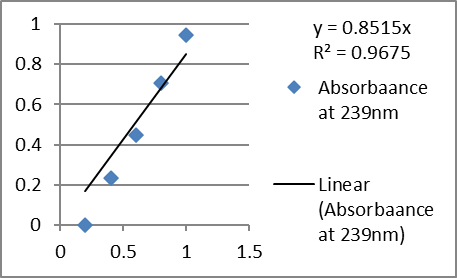
D. Calibration curve in phosphate buffer pH 7.4 at λmax 239nm
3. Phase solubility:
Atovastatin calcium trihydrate was found to be practically insoluble in water, the solubility in different solvent system having different pH was carried out, and results are shown in Table and effect of solvent system shown in figure.
Table: Solubility of Atovastatin calcium in different solvent
|
Media |
Solubility (µg/ml) |
|
Water |
0.005588 |
|
Methanol |
0.006332 |
|
Phosphate buffer pH 6.2 |
0.008790 |
|
Phosphate buffer pH 7.4 |
0.006174 |
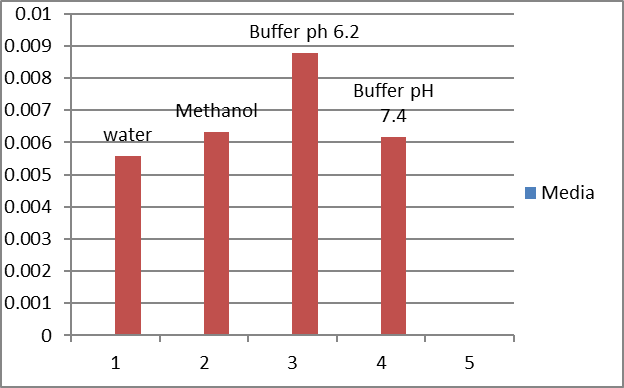
Aovastatin calcium solubility(ug/ml) in different solvent system
4. Assay of selected compact:
|
Test |
Specification |
Result |
|
Assay (HPLC) |
98.0% TO 102.0% |
99.1% |
5. Compact preparation:
|
Sr. No |
Compact 1 |
Compact 2 |
Compact 3 |
Compact 4 |
Compact 5 |
|
1 |
API |
API |
API |
API |
API |
|
2 |
Lactose |
Lactose |
Lactose |
Lactose |
Lactose |
|
3 |
Crospovidone |
Croscarmellose sodium |
Sodium starch glycolate |
Microcrystalline cellulose |
Sodium alginate |
|
4 |
Silica gel |
Silica gel |
Silica gel |
Silica gel |
Silica gel |
|
5 |
MCC |
MCC |
MCC |
MCC |
MCC |
|
6 |
Magnesium stearate |
Magnesium stearate |
Magnesium stearate |
Magnesium stearate |
Magnesium stearate |
From above compact preparation we select Sodium starch glycolate which shows high solubility i.e 90%.
6. FTIR:
As characteristic peaks of the drug were observed it can be concluded that the IR spectrum of drug complies with its chemical structure (Fig ). The characteristic peaks are shown in Table and spectrum is shown in Figure.
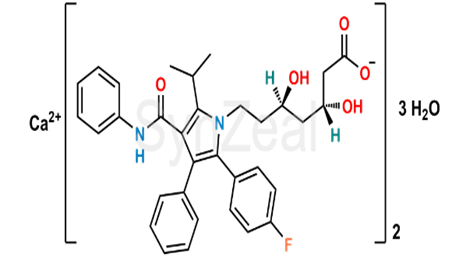
Fig -Structure Of Atovastatin Calcium
Drug 1

Figure: FTIR spectrum of Atovastatin calcium trihyadrate
Drug 2

Figure: FTIR spectrum of Atovastatin calcium Compact
Result :
N-H stretching and C=O stretching at 3364.21 cm-1 and 1649.81 cm-respectively. From the Figure it is evident that inliquisolid compact (B) undergoes no chemical reactionwith any of the excipients used in the preparation ofliquisolid compacts.
7. Differential scanning calorimetric Analysis (DSC)-
Drug :-

Formulation:-

Figure: DSC thermogram of Atovastatin calcium
From above, we conclude that atorvastatin formulation doesn’t go through any structural modification. Hence, it is stable with other excipients without any degradation.
8. PXRD Analysis:
The diffraction pattern of pure Atovastatin calcium trihyadrate shown in figure. It is highly crystalline in nature as indicated by numerous peaks .From the X-ray diffraction peak we conclude that –
The conversion from crystalline to amorphous indicate that the solubility gets increases ; because as compare to drug XRD the complex peaks at a low height.
Drug :-
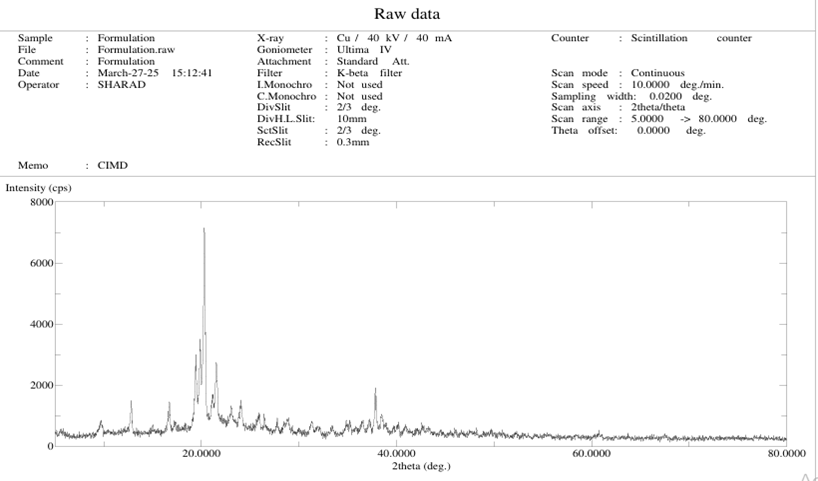
Formulation :-

9. Tablet evaluation :-
|
Liquisolid system (LS) |
Friability % |
Hardness Kg/cm2 |
Weight variation(mg) |
Disintegration time(sec) |
|
1 |
0.56±0.16 |
3.6 |
611.3±0.112 |
40.66 ± 1.15 |
|
2 |
0.61±0.03 |
3.6 |
485.45±0.094 |
41.36 ± 1.10 |
|
3 |
0.594±0.16 |
3.6 |
400.31±0.162 |
42.73 ± 0.64 |
|
4 |
0.632±0.03 |
3.6 |
338.13±0.416 |
46.10 ± 0.17 |
|
5 |
0.503±0.13 |
3.5 |
296.21±0.071 |
47.96 ± 0.95 |
|
6 |
0.666±0.13 |
3.7 |
190.11±0.22 |
48.85 ± 0.51 |
|
7 |
0.598±0.14 |
3.5 |
232.12±0.071 |
50.55 ± 0.78 |
|
8 |
0.599±0.21 |
3.4 |
142.74±0.147 |
51.37 ± 1.07 |
|
9 |
0.654±0.23 |
3.6 |
114.3±0.103 |
53.70 ± 0.61 |
|
10 |
0.549±0.11 |
3.5 |
1.402±0.103 |
55.87 ± 0.52 |
10. In-vitro dissolution studies :-
In- vitro dissolution study for capsules kept for stability were carried out in phosphate buffer pH 6.2 Percent release is mentioned in Table.
|
TIME |
% Drug Release |
|
|
Drug |
Marketed |
|
|
30 |
9 |
10 |
|
60 |
13.32 |
15.55 |
|
90 |
14.07 |
18.88 |
|
120 |
14.9 |
21.67 |
|
150 |
17.82 |
32.15 |
|
180 |
18.9 |
38.12 |
|
210 |
21.33 |
40.12 |
|
240 |
28.55 |
50.88 |
|
270 |
31.62 |
53.55 |
|
300 |
33.43 |
55.76 |
|
330 |
36 |
59.12 |
|
360 |
41 |
61.55 |
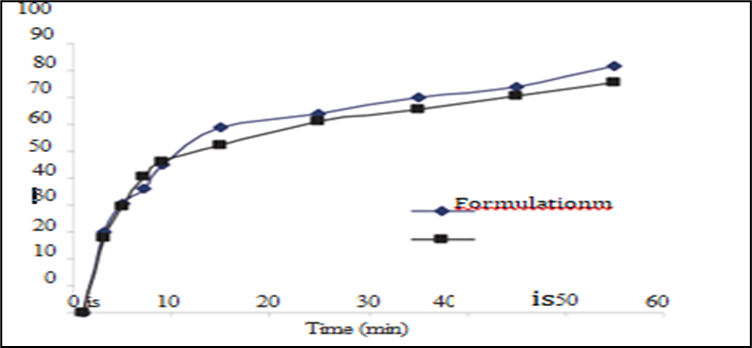
CONCLUSION:-
The present study successfully demonstrates the application of the liquisolid technique as an effective strategy to enhance the solubility and dissolution rate of Atorvastatin Calcium, a BCS Class II drug characterized by poor water solubility and limited oral bioavailability. By formulating Atorvastatin Calcium into liquisolid compacts using non-volatile solvents like propylene glycol and polyethylene glycol 400, along with suitable carrier materials such as Avicel PH 102 and Neusilin US2, significant improvements in dissolution profiles were observed compared to conventional directly compressed tablets.
Characterization studies, including Differential Scanning Calorimetry (DSC) and X-ray Powder Diffraction (XRD), confirmed the transformation of the drug from its crystalline form to an amorphous state within the liquisolid matrix, contributing to enhanced solubility. Furthermore, in vivo bioavailability studies indicated a marked increase in the absorption of Atorvastatin Calcium from the liquisolid formulations, underscoring the technique's potential in improving therapeutic efficacy.
The liquisolid approach also offers advantages in terms of manufacturing, as it allows for the production of tablets with acceptable flow properties and mechanical strength without the need for complex processing techniques. This positions the liquisolid technique as a promising and scalable method for enhancing the oral delivery of poorly soluble drugs like Atorvastatin Calcium.
In conclusion, the liquisolid technique effectively addresses the solubility challenges associated with Atorvastatin Calcium, leading to improved dissolution rates and bioavailability. This novel formulation strategy holds significant promise for the development of more efficacious oral dosage forms of poorly water-soluble drugs.
REFERENCES


P. D. Chaudhari, Priyanka Bagade, Arati Chavan, Bhagwat Bochare, Solubility Enhancement of Atorvastatin Calcium Using Novel Technique, Int. J. of Pharm. Sci., 2025, Vol 3, Issue 9, 2360-2371. https://doi.org/10.5281/zenodo.17168604
 10.5281/zenodo.17168604
10.5281/zenodo.17168604