
We use cookies to ensure our website works properly and to personalise your experience. Cookies policy
Department- Pharmaceutical Chemistry,S. N. College of Pharmacy
A major global public health concern, the rapid rise of antimicrobial resistance (AMR) presents a threat to the efficacy of conventional therapeutic medications and is a factor in the deaths of millions of people around the world, particularly due to the prevalence of ESKAPE bacteria. Bacterial resistance to classical sulfonamides has worsened due to plasmid-mediated resistance acquisition, substrate overproduction, active multidrug efflux mechanisms, target site chromosomal mutations, and the dihydropteroate synthase (DHPS) enzyme in the folic acid biosynthesis pathway.To meet the urgent demand for new antibacterial drugs, this study focuses on the rational design and synthesis of next-generation antimicrobial drugs using a strategic molecular hybridisation method. The research specifically elucidates the synthesis of Atrazine-Modified Sulfonamide analogues, which involve the covalent linkage of a sulfonamide pharmacophore to an s-triazine (atrazine) core. This hybridisation seeks to circumvent proven resistance mechanisms by utilising the improved lipophilicity, structural stiffness, and hydrogen-bonding capabilities of the s-triazine scaffold to augment membrane permeability and intracellular retention, notably against Gram-negative bacteria. We made three new hybrid derivatives, “4-methyl-N-(4-(ethylamino)-6-(isopropylamino)-1,3,5-triazin-2-yl) benzenesulfonamide (3a), 4-methoxy-N-(4-(ethylamino)-6-(isopropylamino)-1,3,5-triazin-2-yl) benzenesulfonamide (3b), and 4-amino-N-(4-(ethylamino)-6-(isopropylamino)-1,3,5-triazin-2-yl) benzenesulfonamide (3c)”. We made these through a very controlled, sequential three-step, temperature-dependent nucleophilic aromatic substitution starting from cyanuric chloride. The synthesised compounds were subjected to thorough physicochemical analysis, encompassing accurate determinations of melting point, yield percentage, and solubility in diverse polar and non-polar organic solvents. Structural validation and chemical purity were clearly validated utilising modern spectroscopic techniques, notably Fourier Transform Infrared (FTIR) spectroscopy, Proton Nuclear Magnetic Resonance (1H-NMR), and Electrospray Ionization Mass Spectrometry (ESI-MS).Structure-Activity Relationship of the research demonstrated that the electronic environment of the para-substituted benzenesulfonamide ring significantly affects its biological and antibacterial efficacy. It was noted that electron-donating groups (EDGs) markedly improved the antibacterial characteristics of the hybrid compounds. Compound 3c, featuring a primary amino group (-NH?), emerged as the most potent and promising lead compound within the synthesized series. Its superior efficacy is directly attributed to its strong electron-donating capability and hydrogen bond donor capacity, which closely mimic the natural bacterial substrate para-aminobenzoic acid (PABA), thereby theoretically maximizing competitive binding affinity within the bacterial DHPS active site. Conversely, the 4-methoxy and 4-methyl derivatives exhibited moderate to lower efficacy, respectively, primarily due to the absence of comparable hydrogen-bonding potential and overall weaker electronic activation. Ultimately, this study confirms the successful synthesis and characterization of highly stable atrazine-modified sulfonamides that hold significant clinical promise for overcoming multidrug-resistant bacterial infections. In addition to their robust scientific potential, these optimized, high-yield synthetic pathways aim to deliver highly targeted therapeutics that reduce the overall required clinical dosages. This subsequently minimizes the accumulation of active pharmaceutical ingredients in wastewater and aquatic ecosystems, addressing the critical ecological concerns associated with environmental antimicrobial resistance
1.1 Background of the Study
The Global Crisis of Antimicrobial Resistance (AMR)
Antimicrobial resistance (AMR) has quickly gone from a possible danger to one of the biggest public health and economic problems in the world in the 21st century. The emergence of antibiotic-resistant strains occurs when prokaryotes (such as bacteria, viruses, fungi, and parasites) evolve to resist conventional medicine. Infections become more difficult, if not impossible, to cure according to this. This silent pandemic is alarming in scale. Bacterial AMR was responsible for 1.14 million fatalities in 2021 and was associated with an additional 4.71 million deaths globally, according to a groundbreaking study published in The Lancet in 2024 by the Global Research on Antimicrobial Resistance (GRAM) Project [1]. The present estimates show that AMR will be directly responsible for roughly 39 million fatalities from 2025 to 2050, with low- and middle-income nations bearing the brunt of this problem [1]. One out of six bacterial infections globally that have been proven in laboratories is now resistant to conventional antibiotic treatments; this trend was further highlighted in the 2025 Global Antibiotic Resistance Surveillance Report by the World Health Organization (WHO). This could undo a century of medical progress [2]. Most often encountered in healthcare contexts are the so-called "ESKAPE" pathogens, which include Enterococcus faecium, Staphylococcus aureus, Klebsiella pneumoniae, Acinetobacter baumannii, Pseudomonas aeruginosa, and other types of Enterobacter. They are very resistant to many drugs (MDR) [3]. There is a lot of worry about gram-negative bacteria because they have strong outer membranes, advanced efflux pumps, and beta-lactamase enzymes. People all across the world are becoming more resistant to third-generation cephalosporins and carbapenems, which were once thought to be strong last-resort treatments. This makes normal empirical remedies useless and raises the costs of healthcare around the world, as well as the number of deaths and illnesses [2].
The Need for Novel Antibacterial Agents:
Consequently, there is an urgent and critical need for the discovery and development of novel antibacterial agents. The current clinical pipeline of antibiotics is widely considered insufficient to combat the rapid emergence of resistant phenotypes. Many newly approved drugs are merely derivatives of existing classes, offering only temporary reprieves before bacteria inevitably develop cross-resistance [4]. To outpace bacterial evolution, modern medicinal chemistry must pivot toward innovative structural interventions that either target entirely new biological pathways or effectively bypass established resistance mechanisms.
One highly effective strategy to overcome these limitations is the molecular hybridization approach, which involves the amalgamation of two or more distinct pharmacophores into a single molecular entity [5]. Sulfonamides, despite being one of the oldest classes of synthetic antimicrobials, remain a highly valuable structural core due to their well-established mechanism of inhibiting bacterial folic acid synthesis. However, their modern clinical utility has been severely compromised by widespread microbial resistance. By chemically modifying the sulfonamide core with an s-triazine (atrazine) moiety, a versatile scaffold known for its broad spectrum of biological activities and ability to engage multiple molecular targets it is hypothesized that the resulting hybrid molecules would exhibit synergistic antibacterial efficacy. This novel structural hybridization aims to restore sensitivity against resistant bacterial strains and present a reduced propensity for future resistance development, directly addressing the critical need for next-generation antimicrobials.
1.2 Sulfonamides as Antibacterial Agents
The escalating crisis of antimicrobial resistance necessitates not only the discovery of novel chemical classes but also the strategic revitalization of established, well-tolerated pharmacophores. Among these, sulfonamides hold a distinguished place. Introduced in the 1930s following Gerhard Domagk’s Nobel Prize-winning discovery of the prodrug Prontosil, sulfonamides represent the first major class of synthetic, systemically active antibacterial agents [6]. Their introduction revolutionized modern medicine, drastically reducing mortality rates from previously fatal streptococcal and staphylococcal infections and laying the foundational principles of modern antimicrobial chemotherapy. Because of their synthetic tractability, broad spectrum of activity, and favourable pharmacokinetic profiles, sulfonamides remain a cornerstone in medicinal chemistry. However, to maintain their clinical relevance against modern multidrug-resistant pathogens, their traditional structures must be innovatively modified.
1.2.1 Mechanism of Action: Inhibition of Folic Acid Synthesis
The therapeutic success of sulfonamides is rooted in their selective toxicity, a principle driven by the fundamental metabolic differences between bacterial and mammalian cells. Mammals absorb preformed folate from their diet, whereas most bacteria must synthesize their own folic acid de novo to produce the purines and pyrimidines necessary for DNA and RNA replication.
Sulfonamides act as bacteriostatic agents by disrupting this essential biosynthetic pathway. Structurally, the sulfonamide pharmacophore is a close analogue of paminobenzoic acid (PABA), a natural precursor in the bacterial synthesis of folic acid. When PABA and pteridine are typically catalysed to produce dihydropteroic acid, the enzyme dihydropteroate synthase (DHPS) is competitively inhibited by sulfonamides [7]. By overwhelming the active site of DHPS, sulfonamides stall the production of downstream folates, ultimately halting bacterial cell growth and division.
1.2.2 Current Limitations and Mechanisms of Resistance
Despite their elegant mechanism of action, the clinical utility of traditional sulfonamides (such as sulfamethoxazole and sulfadiazine) has been severely eroded by decades of extensive use in both human medicine and agriculture. Bacteria have evolved highly efficient, multifaceted strategies to circumvent sulfonamide-induced inhibition:
1.2.3 The Rationale for Structural Modification
Because pathogenic bacteria have largely adapted to the classical, unmodified sulfonamide structure, there is a compelling mandate to design next-generation derivatives. By integrating the sulfonamide pharmacophore with an atrazine (s-triazine) scaffold, a molecule recognized for its ability to independently permeate bacterial membranes and interact with diverse biological targets, it is possible to create a hybrid molecule. This hybridization strategy aims to bypass established resistance mechanisms, enhance cellular uptake, and potentially introduce secondary mechanisms of action, thereby restoring and amplifying the antibacterial activity of the sulfonamide core.
1.3 The Role of s-Triazines (Atrazine)
In the pursuit of novel antimicrobial agents to combat multidrug-resistant pathogens, heterocyclic compounds heavily dominate modern drug discovery. The s-triazine (1,3,5-triazine) ring stands out among various structural patterns; it is a six-membered aromatic system with three nitrogen atoms placed symmetrically. The unique arrangement of heteroatoms in the s-triazine core imparts significant physicochemical advantages, including enhanced hydrogen-bonding capabilities, structural rigidity, and favourable lipophilicity profiles, all of which are critical for facilitating interactions with complex bacterial drug targets [10].
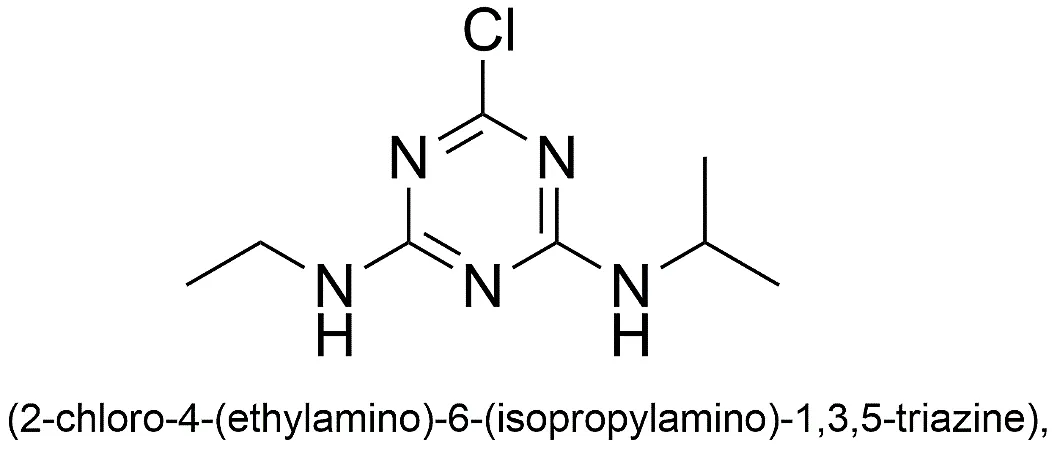
Fig.1.1: Representation of the chemical structure of Atrazine.
1.3.1 Cyanuric Chloride
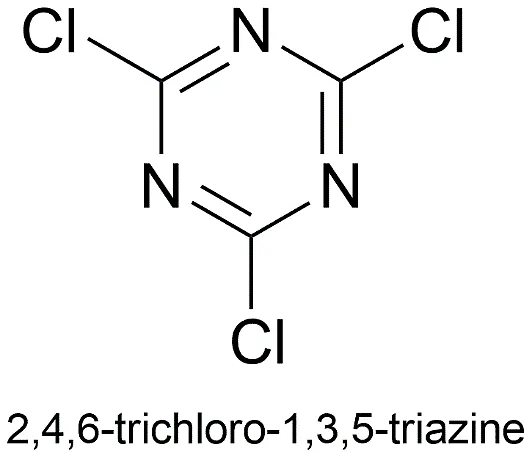
Fig.1.2: Representation of the chemical structure of Cyanuric chloride
The significant importance of s-triazine in medicinal chemistry is mostly attributable to the remarkable synthetic usefulness of its precursor, 2,4,6-trichloro-1,3,5-triazine (cyanuric chloride). Cyanuric chloride is a cheap starting material that can be bought in stores. It makes it possible to quickly, precisely, and in many ways make things. The three chlorine atoms on the triazine ring react differently to nucleophilic substitution (such with amines, alcohols, and thiols) depending on the temperature. Typically, the first substitution takes place between 0 and 5 °C, the second between 20 and 25 °C, and the third between 80 and 100 °C. [11].
The step-wise regioselectivity of the s-triazine core makes it a great modular scaffold for combinatorial chemistry and the smart construction of complex hybrid compounds. Atrazine (2-chloro-4-(ethylamino)-6-(isopropylamino)-1,3,5-triazine), conventionally employed as a herbicide, illustrates this particular substitution sequence. Its highly stable framework and ability to independently cross biological membranes make modified atrazine derivatives highly attractive starting points for pharmaceutical repurposing.
1.3.2 Pharmacological Importance and Antibacterial Potential
Derivatives derived from the s-triazine scaffold demonstrate an exceptionally extensive range of pharmacological activity, encompassing anticancer, antimalarial, antiviral, and, most significantly, robust antimicrobial properties [12]. When designing antibacterial drugs, s-triazines are known to work in ways that are different from those of standard antibiotics. Numerous triazine derivatives have been reported to intercalate with bacterial DNA, compromise outer membrane integrity, and inhibit critical bacterial enzymes, notably dihydrofolate reductase (DHFR), which operates downstream of dihydropteroate synthase (DHPS) in the bacterial folic acid pathway [13].
Because the s-triazine core is capable of engaging multiple biological targets simultaneously, it significantly reduces the probability of bacteria developing rapid target-based resistance. By leveraging the lipophilic and hydrogen-bonding characteristics of the atrazine-type scaffold, medicinal chemists can optimize the pharmacokinetic properties of attached pharmacophores, ensuring higher intracellular accumulation within pathogenic bacteria. This versatility provides a powerful rationale for utilizing the atrazine framework. When strategically coupled with established antibacterial moieties that are currently failing due to resistance, the s-triazine core acts not just as a structural linker, but as an active participant in restoring and amplifying therapeutic efficacy.
1.4 The Concept of Molecular Hybridization
To combat the multifactorial nature of antimicrobial resistance, modern medicinal chemistry has increasingly shifted away from the traditional "one-target, one-drug" paradigm. As an alternative, researchers are turning to molecular hybridisation, a relatively new but very effective method of drug design that forms new chemical entities by covalently combining two or more bioactive pharmacophores. [5]. The primary objective of this approach is to produce hybrid molecules that exhibit synergistic or dual-action properties, offering enhanced biological activity and improved pharmacokinetic profiles compared to the individual parent compounds [14]. By presenting a completely new structural motif to bacterial cells, hybrid molecules can simultaneously engage multiple biological targets, significantly reducing the likelihood of cross-resistance and target mutation.
1.4.1 Rationale for the Atrazine-Sulfonamide Hybrid
The combination of an atrazine (s-triazine) core with a sulfonamide pharmacophore represents a highly rational and strategic application of molecular hybridization. As previously established, traditional sulfonamides are highly specific inhibitors of dihydropteroate synthase (DHPS), but their clinical efficacy is largely compromised by plasmid-mediated resistance and altered cellular permeability. The hybridization strategy seeks to rescue the therapeutic value of the sulfonamide class by utilizing the s-triazine scaffold as a potent biological anchor and permeability enhancer.
Recent advances in multicomponent drug design have highlighted that linking sulfonamides with complementary heterocyclic rings, such as triazines, generates hybrid structures capable of overcoming established drug resistance [15]. The symmetrical, electron-deficient s-triazine core provides excellent hydrogen-bonding capabilities and structural rigidity, which are vital for strong target enzyme affinity [16]. Furthermore, the lipophilic side chains inherent in the atrazine derivative facilitate superior penetration through the complex lipid bilayers of both Gram-positive and Gram-negative bacteria. Once inside the cell, the hybrid molecule is assumed to act synergistically: the s-triazine moiety may disrupt cellular membranes or secondary enzymatic targets (such as dihydrofolate reductase), while the sulfonamide moiety independently blocks folic acid synthesis.
1.4.2 Transition to the Research Problem
Ultimately, the hybridization of atrazine and sulfonamide bridges the gap between historical drug utility and modern clinical necessity. While pathogenic bacteria have adapted to recognize and expel classic sulfa drugs, the introduction of the bulky, multi-functional atrazine scaffold creates a steric and electronic profile that bacterial efflux pumps and mutated DHPS enzymes are unlikely to recognize. This sophisticated chemical modification transitions directly into the core problem addressed by this study: standard monotherapies are failing, and there is a critical need to design, synthesize, and evaluate new atrazine-modified sulfonamides to restore and enhance antibacterial activity against resistant strains.
1.4.3 Overcoming Target-Site Mutational Resistance
The necessity for this specific structural modification—covalently linking an atrazine (s-triazine) core to a sulfonamide pharmacophore, is driven by the precise molecular mechanisms of modern bacterial resistance. Traditional sulfonamides are structurally streamlined to mimic p-aminobenzoic acid (PABA), allowing them to competitively bind the narrow active site of dihydropteroate synthase (DHPS). However, clinically relevant pathogens have acquired plasmid-encoded sul genes that express mutated DHPS variants. These mutated enzymes exhibit altered active-site topographies that sterically exclude classic sulfonamides while retaining affinity for PABA [17]. Modifying the sulfonamide with a bulky, electron-deficient s-triazine scaffold addresses this specific limitation. The introduction of the atrazine moiety radically alters the spatial geometry, steric bulk, and electrostatic surface area of the molecule. This specific hybridization is designed to force the molecule into secondary binding interactions within or adjacent to the DHPS active site, overcoming the localized point mutations that currently render standard therapies ineffective [18].
1.4.3 Bypassing Efflux and Enhancing Permeability
Furthermore, a primary mechanism of multidrug resistance in Gram-negative bacteria is the upregulation of non-specific efflux pumps, which actively extrude classic, low-molecular-weight antibiotics before they can reach therapeutic intracellular concentrations. The specific selection of the atrazine core is strategically necessary to counteract this. The s-triazine ring, particularly when substituted with alkylamine groups (as seen in atrazine), significantly increases the overall lipophilicity (log P) of the hybrid molecule [19]. This enhanced lipophilic character is essential for facilitating superior passive diffusion across the complex lipopolysaccharide outer membrane of Gram-negative strains. Once internalized, the altered molecular weight and distinct structural motif of the hybrid are hypothesized to be poorly recognized by standard bacterial efflux machinery, thereby increasing intracellular retention times [19].
1.4.4 Methodological Justification
Because the exact relationship between the hybrid's structure and its biological activity cannot be universally predicted, an empirical, systematic synthetic methodology is strictly necessary. The proposed approach—generating distinct, incrementally modified sets of derivatives (such as the planned 3-series and 5-series compounds)—is essential to isolate and evaluate the distinct electronic and steric contributions of various functional groups attached to the hybrid core.
Validating that this specific molecular hybridization has been successfully achieved requires stringent analytical documentation. The precise structural integrity of the atrazine-sulfonamide linkage must be confirmed through targeted mass spectrometry to verify exact molecular mass shifts, alongside rigorous 1H-NMR spectroscopy to map the chemical environments of the newly formed bonds. By confirming these structures with high analytical precision before biological screening, this methodology ensures that any observed enhancement in antibacterial activity can be definitively attributed to the specific atrazine-modified sulfonamide architecture.
1.5 Sulfonamides and 1,3,5-Triazine Derivatives
Sulfonamides, characterized by the presence of the sulfonyl group attached to an amine (SO2NH2), represent the first class of synthetic antibacterial agents successfully used in clinical medicine. While traditionally derived from coal tar dyes like Prontosil, modern medicinal chemistry focuses on the development of structural analogues using "privileged scaffolds" such as the 1,3,5-triazine ring. This nitrogen-rich heterocyclic core is most notably recognized in the herbicide Atrazine a widely used agrochemical for weed control in maize and sorghum [20]. The white crystalline triazine core is a flexible "model" for molecular hybridisation and looks like a solid. By substituting the chlorine and alkylamino groups found in Atrazine with sulfonamide moieties, researchers can engineer molecules with enhanced affinity for bacterial enzymes. The integration of a sulfonamide onto the triazine scaffold targets the folic acid biosynthesis pathway, specifically inhibiting the enzyme dihydropteroate synthase (DHPS). Beyond their primary antibacterial role, these modified derivatives are being investigated for their potential to overcome antibiotic resistance [21]
1.5.1 Chemistry of Sulfonamide
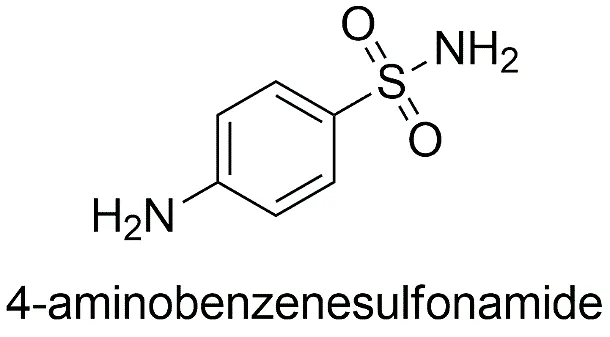
Fig.1.3: Representation of the chemical structure of Sulfonamide.
1.5.2 Chemistry of 1,3,5 Triazine
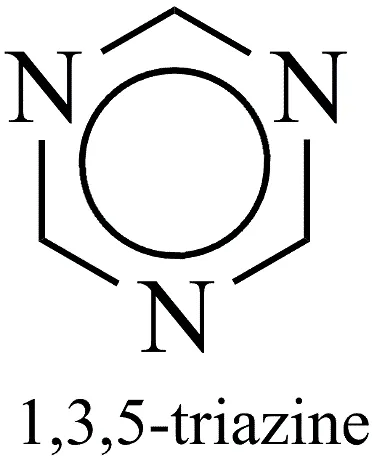
Fig.1.4: Representation of the chemical structure of 1,3,5-triazine.
1.6 AIM AND OBJECTIVES
Aim: The aim of my present work is “Development of Atrazine Modified Sulfonamide for Enhanced Antibacterial Activity”, which was fulfilled by following objectives:
Objectives:
REVIEW OF LITERATURE
2. 1 Description of Sulfonamide
Many researchers have examined sulfonamides due to their chemical nature as synthetic antimicrobial agents, characterized by a sulfamoyl group (-SO₂NH₂) and a modifiable amino group (-NH₂) combined to a benzene ring, which confer unique chemical reactivity and biological activity. Its structure allows it to act as an antibacterial agent by competitively inhibiting dihydropteroate synthase to block bacterial folate synthesis and its nucleophilic properties allow it to participate in a variety of chemical reactions—specifically positional modification via regioselective substitution at the C6 position of an atrazine core—making it an important compound in studies on molecular hybridization, synthetic intermediates, and overcoming microbial resistance [17].
Table 2.1 Drug Profile of Sulfonamide
|
S. No. |
Property |
Description |
|
1. |
Drug |
Sulfonamide |
|
2. |
Molecular Formula |
C6H₈N2O2S |
|
3. |
Molecular Weight |
172.21 g/mol |
|
4. |
Elemental Composition |
C: 47.7%, H: 4.4%, N:16.6%, O: 18.9%, S:12.6% |
|
5. |
Preparation Method |
Reduction of p-nitrobenzenesulfonamide using agents like nano-iron powder, magnesium, or zinc in alcohol solvents to prepare specialized derivatives. |
|
6. |
IUPAC Name |
4-aminobenzenesulfonamide |
|
7. |
Odour |
Odourless |
|
8. |
Colour |
White |
|
9. |
Solubility |
Acetone, Alcohol, Methanol, Ethanol, sparingly soluble in water |
|
10. |
Melting Point |
165°C |
|
11. |
Synonyms |
Sulfonamide, Sulphonamide, 4-Aminobenzenesulfonamide, Sulfanilamide |
|
12. |
Vapor Pressure |
0.0000073 [mmHg] |
|
13. |
pKa |
10.6 (at 20 °C) |
|
14. |
Log P |
-0.62 |
|
15. |
Structure |

|
2.2 Review on Current trends and the biological mechanisms of bacterial resistance
Christopher J. L. Murray et al. [22]: Estimates the global burden of bacterial antimicrobial resistance, revealing that AMR was a leading cause of death worldwide in 2019, emphasizing the urgent need for novel interventions to counteract this global crisis.
Wanda C. Reygaert et al. [23]: Bacterial resistance and its molecular mechanisms, including the ways in which bacteria use multidrug active efflux pumps, change drug targets, enzymatically inactivate drugs, and reduce drug absorption, prevent therapeutic interventions.
Giuseppe Mancuso et al. [24]: Investigates the current trends in antibiotic resistance among the most critical clinical pathogens, particularly focusing on the ESKAPE group and their sophisticated biological ability to escape the biocidal action of standard antimicrobial agents.
Jessica M. A. Blair et al. [25]: The molecular and genetic basis of antibiotic resistance, detailing how intrinsic and acquired resistance mechanisms, including the horizontal transfer of resistance genes via plasmids, compromise the clinical efficacy of major antibiotic classes.
Francesca Prestinaci et al. [26] It describes about the antimicrobial resistance as a global multifaceted phenomenon, discussing the epidemiological trends, the profound socioeconomic impact, and how the widespread misuse of antibiotics directly accelerates the evolutionary mechanisms of bacterial resistance.
Jose M. Munita et al. [27]: Complex biochemical mechanisms of antibiotic resistance, providing a rigorous classification of how bacteria strategically alter the structural conformation of their target sites to reduce drug affinity while maintaining essential physiological functions.
D. M. De Oliveira et al. [3]: The specific resistance mechanisms employed by high-priority multidrug-resistant pathogens, highlighting how these bacteria synergistically combine outer membrane impermeability, upregulated efflux pumps, and target mutation to achieve extensive drug-resistant profiles.
2.3 Review on SAR of classic Sulfanilamide and recent advancements in Sulfonamide derivatives
Zafar et al. [28]: Explores the synthetic processes and historical relevance of classic sulfa drugs and investigates the structure-activity relationship (SAR), emphasizing the absolute necessity of the free para-amino group (N4) and the direct attachment of the sulfonamide group to the benzene ring for the competitive inhibition of bacterial dihydropteroate synthase (DHPS).
Oudah et al. [6]: Discovery of novel sulfonamides as antibacterial agents and discusses the critical shift from traditional monotherapies toward the development of dual-action hybrid derivatives to overcome widespread bacterial resistance, detailing how structurally modified sulfonamides retain their DHPS-inhibiting core while engaging secondary bacterial targets.
C. Capasso et al. [7]: Investigates the specific biochemical mechanisms of sulfa drugs as antimetabolites and van der Waals interactions and hydrogen-bonding requirements within the DHPS active site, further elaborating on how N1 substitutions with heterocyclic rings improve the ionization state to closely mimic the natural substrate, p-aminobenzoic acid (PABA).
R. Ghomashi et al. [29]: It describes about recent advances in biologically active multicomponent sulfonamide hybrids. Demonstrate that integrating the sulfonamide pharmacophore with distinct, electron-deficient heterocyclic rings (such as triazines) represents a highly effective modern advancement for bypassing established target-site resistance and efflux pump mechanisms.
M. J. Mengelers et al. [30]: Examines the crucial relationship between physicochemical properties and the antibacterial activity of sulfonamides. The study provides empirical data showing that optimizing the hydrophobicity and acid dissociation constant (pKa) through specific structural modifications is critical for penetrating the complex outer cellular membranes of Gram-negative pathogens.
O. Sköld et al. [8]: The exact biological mechanisms rendering classic sulfonamides ineffective, specifically detailing the mutational alterations in chromosomal folP genes and plasmid-borne sul genes and provides the biological validation for why modern advancements in sulfonamide chemistry must hybrid functional groups to successfully bypass these altered DHPS active sites.
Liu W. et al. [31]: Explain about recent synthetic methodologies for generating novel sulfonamide derivatives and focuses on the optimization of reaction conditions to improve the yield and purity of sulfonamide conjugates, highlighting how analytical characterization techniques like; NMR and mass spectrometry are essential for verifying the successful linkage of new pharmacophores to the sulfonamide core.
2.4 Review on Synthesis Strategies for Modifying Cyanuric Chloride/Atrazine
G. Blotny [32]: Describes the extensive applications of 2,4,6-trichloro-1,3,5-triazine (cyanuric chloride) in organic synthesis and comprehensively explains the highly predictable, temperature-controlled, stepwise nucleophilic aromatic substitution of the three equivalent chlorine atoms (occurring sequentially at 0 °C, room temperature, and elevated reflux temperatures), making it a privileged, modular building block for hybrid drug design.
M. I. Ali et al. [33]: Producing complex heterocyclic hybrids from cyanuric chloride. And highlight the synthetic tractability and high reactivity of the s-triazine core, demonstrating how medicinal chemists can sequentially install diverse biological pharmacophores on a single nucleus, thereby tailoring specific lipophilic profiles akin to the atrazine scaffold.
Anupama et al. [34]: Synthesis of biologically active s-triazine-based multi-component molecules and provides detailed synthetic protocols for utilizing the temperature-dependent differential reactivity of cyanuric chloride to successfully append multiple distinct nucleophiles, validating the methodology required to bridge independent pharmacophores into a single hybrid entity.
X. Y. Liu et al. [35]: Systematically examines the potent antimicrobial and antifungal potential of s-triazine derivatives and demonstrate that the rigid, nitrogen-rich planar structure of s-triazines is highly effective at engaging secondary cellular targets and disrupting pathogenic membranes, making it an exceptional framework for developing next-generation antimicrobial agents.
K. Sharma et al. [36]: Explores the broad biological profile of the 1,3,5-triazine pharmacophore and how specific substitutions at the 2, 4, and 6 positions, structurally mimicking the architecture of the herbicide atrazine, yield drug candidates with significant antibacterial, antimalarial, and anticancer properties by ensuring high intracellular accumulation and strong hydrogen-bonding with target enzymes.
R. V. Patel et al. [37]: Synthesis and in vitro antibacterial evaluation of tailored s-triazine derivatives and stablished a clear structure-activity relationship (SAR), proving empirically that the strategic placement of specific amines and electron-withdrawing groups on the triazine core significantly enhances biocidal activity against resistant Gram-negative and Gram-positive bacterial strains.
M. A. Alelaimat et al. [16]: The biological profiling of novel sulfonamide–triazine hybrid derivatives. Utilized molecular docking and in vitro screening to prove that covalently combining these two distinct moieties creates a highly stable, biologically active multi-target hybrid capable of overcoming the limitations of standard monotherapies.
2.5 Review on Previously Reported Triazine-Sulfonamide Hybrids
M. A. Alelaimat et al. [16]: Synthesis of a distinctive sequence of sulfonamide-triazine hybrid compounds through successive nucleophilic aromatic substitution of cyanuric chloride via solvent-free fusion techniques. The authors verified that the structural amalgamation of the sulfonamide pharmacophore with an electron-deficient triazine core substantially improves target binding, hence demonstrating the efficacy of this particular hybridisation strategy.
R. Ghomashi et al. [29]: Investigate recent advancements in hybridizing the sulfonamide core with biologically active heterocycles, specifically highlighting triazines. Notes that while generic triazine-sulfonamide combinations demonstrate synergistic biological promise, the specific steric tuning of the triazine substituents to maximize cellular permeability is frequently underexploredR. P. Modh et al. [38]: Evaluated the design, synthesis, and in vitro antimicrobial activity of novel hybrid triazine derivatives. The study established that incorporating the s-triazine scaffold alongside secondary pharmacophores significantly improved the antibacterial and antifungal efficacy against resistant strains, largely due to the rigid, nitrogen-rich core facilitating strong hydrogen-bonding interactions with pathogenic targets.
MATERIALS AND METHODOLOGY
3.1 CHEMICALS, INSTRUMENTS AND APPARATUS REQUIRED
Table 3.1.1 List of chemicals.
|
Chemicals |
Specification / Manufacturer |
|
Cyanuric chloride (2,4,6-Trichloro-1,3,5-triazine) |
Sigma-Aldrich |
|
Isopropylamine |
CDH |
|
Ethylamine |
SRL |
|
Substituted sulfonamides (4-methylbenzenesulfonamide) |
Loba Chemie |
|
Tetrahydrofuran (THF) |
Sigma-Aldrich |
|
1,4-Dioxane |
SRL |
|
Ethanol |
Sigma-Aldrich |
|
Potassium carbonate (Anhydrous K2CO3) |
Sigma-Aldrich |
|
Sodium hydroxide (NaOH) pellets |
Loba Chemie |
|
Dichloromethane (DCM) |
Loba Chemie |
|
Hexane |
CDH |
|
Ethyl acetate |
CDH |
|
Methanol |
Loba Chemie |
|
Deuterated Dimethyl Sulfoxide |
Sigma-Aldrich |
|
Dichloromethane |
Sigma-Aldrich |
|
Silica gel 60 F254 TLC plates |
Sigma-Aldrich |
|
Distilled Water |
Institutional Supply |
|
Acetic acid |
CDH |
|
Acetone |
CDH |
|
DMSO |
Merck |
|
Acetonitrile |
CDH |
|
Isopropyl alcohol |
CDH |
|
Benzene |
Merck |
3.1.2 Instruments
Table 3.1.2 List of Instruments.
|
Instruments |
Source |
|
Analytical Balance |
Vibra (Essae) |
|
Magnetic Stirrer |
A and T scientific industries |
|
Hot Air Oven |
A and T scientific industries |
|
FT-IR Analyzer |
PerkinElmer Spectrum-2 |
|
NMR Analyzer |
Bruker Avance 400 / Avance III HD |
|
Mass Analyzer |
Waters Alliance e2695 / HPLC TQD Mass spectrometer |
|
Vacuum Pump |
VALUE |
|
Refrigerator |
Videocon |
|
Hot Plate |
Skybound |
|
Melting point apparatus |
Contemp |
3.1.3 Apparatus
Research work was carried out and successfully completed utilizing a range of instruments.
Table 3.1.3 List of Apparatus.
|
Round Bottom Flask |
|
Glass Rod |
|
Conical Flask |
|
Funnel |
|
Beaker |
|
Condenser |
|
Thermometer |
|
Burette Stand |
|
Capillary Tube |
|
Pipette |
|
TLC Plate |
|
Volumetric Flask |
|
Micro Plate |
|
Magnetic stirrers |
|
Tripod Stand |
|
Filter Paper |
|
Petri Dish |
|
Separating funnel |
3.2 METHOD
3.2.1 Determination of Melting Point
The melting point was utilized as a standard measure for assessing structural changes and the purity of the organic compounds. A capillary tube was filled with a small quantity of the dry, finely powdered sample of the synthesized atrazine-modified sulfonamide derivative. The tube was then put in a digital melting point machine and slowly heated up. We took record of the actual point that the sample began to melt. At this point, the melting range began. After that, the melting range was crossed when the sample became completely liquid after a 2 to 3 °C per minute temperature increase. It was important to write down both the starting and ending temperatures. Because pure substances typically exhibit a sharp, narrow melting temperature range of 1-3 °C, these recorded values served to verify the purity of the synthesized compounds, as the presence of any impurities would broaden this range. Upon completion of each measurement, the apparatus was cleaned thoroughly to prevent cross-contamination during subsequent analyses [39].
3.2.2 Determination of Solubility
To determine the solubility profile of the synthesized atrazine-modified sulfonamide derivatives, a small, precisely measured quantity of each compound (10 mg) was introduced into 10 mL of a specified test solvent within a test tube. The mixtures were maintained at a constant ambient temperature and agitated to facilitate dissolution. The solvents utilized for this solubility screening encompassed a range of polarities, including acetone, methanol, ethanol, chloroform, carbon tetrachloride, dimethyl sulfoxide (DMSO), and distilled water. The extent of dissolution was visually assessed and recorded to establish the solubility characteristics of the synthesized hybrids, which was critical for selecting the appropriate vehicle solvents for subsequent spectral analysis and in vitro biological evaluations [40].
3.2.3 Determination of Percentage Yield
The percentage yield was utilized as a fundamental calculation to determine the overall efficiency of the chemical reactions involved in synthesizing the atrazine-modified sulfonamide derivatives. To find the percentage yield, divide the practical yield by the theoretical yield. We got this result by comparing the practical yield, which is the actual mass of the purified product we got in the lab, with the theoretical yield, which is the maximum possible mass of the product based on stoichiometric calculations of the limiting reagent. This measurement was crucial for assessing reaction efficiency, optimizing the synthetic pathways, and evaluating resource utilization throughout the study. The percentage yield was calculated using Equation 3.1[39].
Equation (3.1) can be used to calculate the Percentage Yield as:
% Yield=Practical Yield ÷Theorectical Yield×100
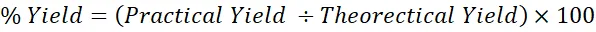
3.2.4 Determination of Rf Values
Thin Layer Chromatography (TLC) was employed to monitor the progress of the chemical reactions and to definitively ascertain the purity of the synthesized atrazine-modified sulfonamide derivatives. The stationary phase was made up of aluminium plates that had been pre-coated with silica gel 60F (254). Using a microcapillary tube, a small amount of the dissolved substance was put on the TLC plate as a separate spot toward the bottom. The spotted plate was then put into a chromatography chamber that had already been equilibrated and had a mobile phase solvent system that worked best with certain amounts of hexane and ethyl acetate. With the lid sealed, capillary action lifted the solvent up the plate. Upon reaching a specific distance, the plate was removed, allowed to dry in the air, and checked for any damage. The distinct compound spots were visualized using a dual-wavelength ultraviolet (UV) cabinet (at 254 nm and 365 nm) and further confirmed by exposure to an iodine vapor chamber. The retention factor (Rf) value, representing the relative mobility of the compound within the selected solvent system, was calculated using Equation (3.2)
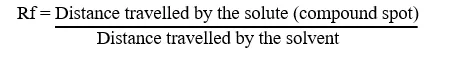
The calculated Rf values, were recorded as specific physicochemical identifiers for each synthesized derivative (e.g., compounds 3a, 3b, and 3c), with the presence of a single, well-defined spot confirming the purity of the final hybridized products [41].
3.3 SYNTHESIS METHODOLOGY
The structural modification of the 1,3,5-triazine core to form atrazine-modified sulfonamide hybrids was strategically designed to combat antimicrobial resistance (AMR). The precursor, cyanuric chloride (2,4,6-trichloro-1,3,5-triazine), was selected due to the highly predictable, temperature-dependent nucleophilic aromatic substitution of its three equivalent chlorine atoms. To enhance the antibacterial activity, the triazine ring was specifically modified at positions 4 and 6 with isopropylamine and ethylamine groups, respectively, synthesizing the "atrazine core." This specific combination of alkyl chains was incorporated to optimize the lipophilicity and membrane permeability of the molecule. Gram-negative bacteria possess a highly restrictive, lipid-rich outer membrane that typically excludes large or polar antibiotics. The enhanced lipophilicity of the atrazine scaffold facilitated superior penetration through these bacterial porins and lipid bilayers.
Subsequently, the remaining chlorine atom at position 2 of the triazine core was substituted with a targeted sulfonamide moiety. The sulfonamide group is a well-known pharmacophore that stops the bacterial enzyme dihydropteroate synthase (DHPS) from working, which stops the production of folic acid. By covalently linking the sulfonamide to the large, nitrogen-rich atrazine core, the hybrid molecule was designed to get beyond the steric hindrance of mutant DHPS enzymes that are seen in resistant strains. The planar triazine ring acted as a bioisostere, providing additional hydrogen-bonding sites (via its nitrogen atoms) to anchor the molecule within the enzymatic active site, thereby significantly enhancing the overall biocidal efficacy compared to standard monotherapies.
3.3.1 General Methodology Synthesis of Atrazine-Modified Sulfonamide Derivatives
The synthesis of the target hybrid compounds was executed through a highly controlled, three-step sequential pathway utilizing the temperature-dependent reactivity of cyanuric chloride.
Step 1: Synthesis of 2,4-dichloro-6-(isopropylamino)-1,3,5-triazine
To begin the procedure 30 mL of analytical-grade acetone was mixed with 0.01 mol of cyanuric chloride (2,4,6-trichloro-1,3,5-triazine). The flask was put in an ice-salt bath to keep the temperature between 0 and 5 °C. An equal amount of 0.01 mol of isopropylamine dissolved in 10 mL of distilled water was added drop by drop to the mixture that was being stirred for 30 minutes. When hydrochloric acid (HCl) was made, a 10% aqueous sodium hydroxide (NaOH) solution was added at the same time to keep the pH at a slightly alkaline level (pH ~8). At a temperature of 0 to 5 °C, the reaction mixture was stirred all the time for two hours. Thus white stuff that came out had been filtered, rinsed with ice, and then dried.
Reaction:
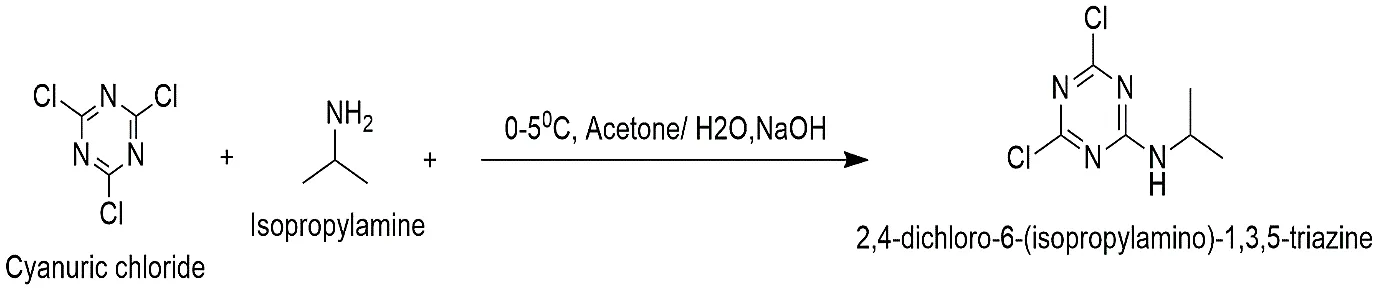
Fig.2.1: Representation of the chemical structure of 2,4-dichloro-6-(isopropylamino)-1,3,5-triazine
Step 2: Synthesis of 2-chloro-4-(ethylamino)-6-(isopropylamino)-1,3,5-triazine (The Atrazine Core)
Dissolved the mono-substituted intermediate from Step 1 (0.01 mol) in 30 mL of tetrahydrofuran (THF). We added ethylamine (0.01 mol) to this solution drop by drop. This second nucleophilic substitution took place at room temperature, which is between 20 and 25 °C. The reaction mixture was agitated hard for 4 to 6 hours, then 10% NaOH was added to keep the pH level alkaline. The mixture was filtered, rinsed with cold water, dried, and then recrystallised from ethanol after being placed onto crushed ice.
Reaction:
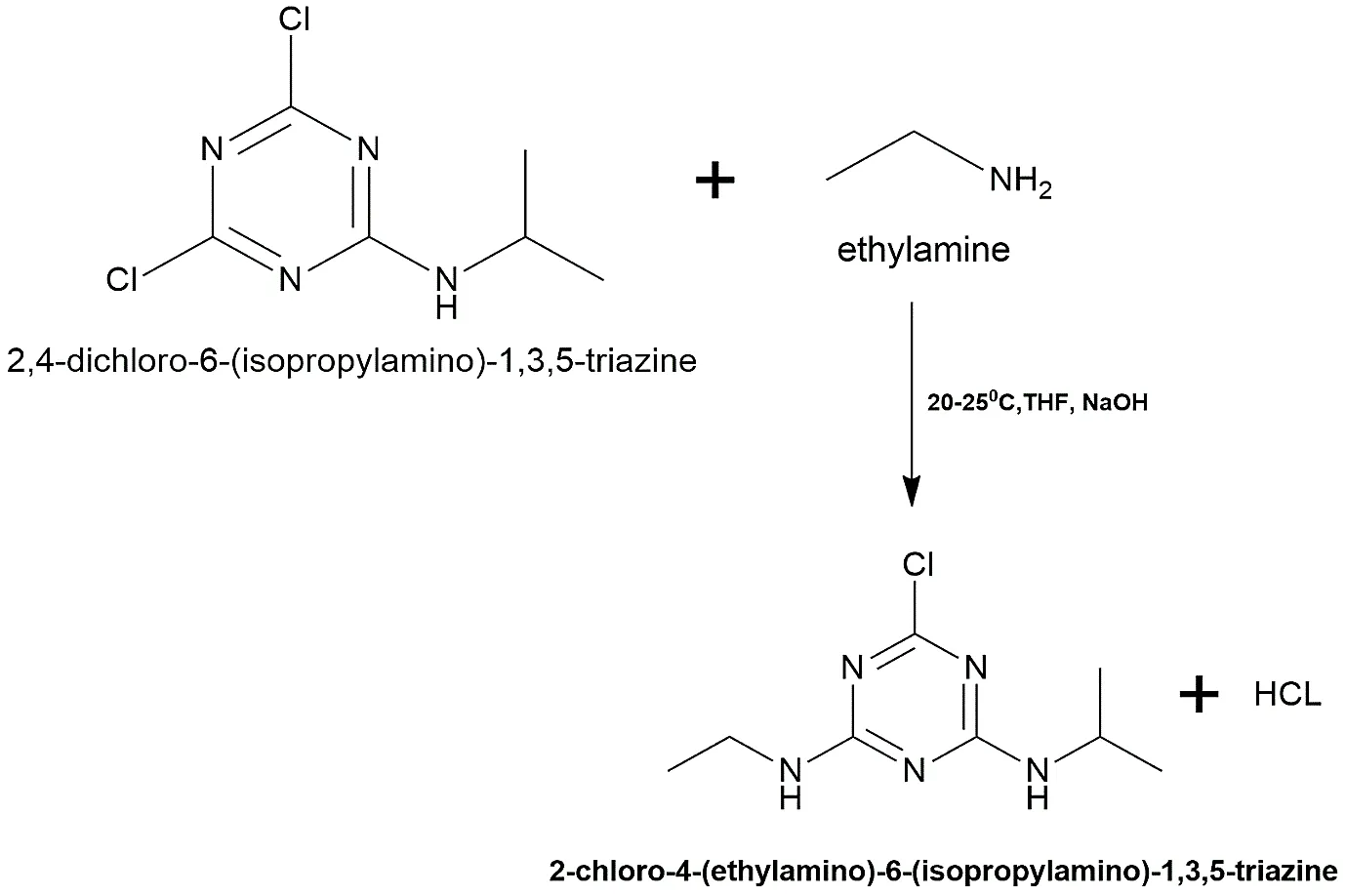
Fig.2.2: Representation of the chemical structure of 2-chloro-4-(ethylamino)-6-(isopropylamino)-1,3,5-triazine
Step 3: Synthesis of the Final Hybrid Compound
In the last phase of the synthesis, 25 mL of 1,4-dioxane was used to dissolve the purified atrazine core from phase 2 (0.005 mol) and the chosen substituted sulfonamide derivative (0.005 mol). We added anhydrous potassium carbonate (K2CO3, 0.005 mol) to soak up the acid. For 12 to 18 hours, the reaction mixture was heated to 80–100 °C and stirred vigorously. After TLC confirmed the results, the solvent was concentrated, and the residue was added to ice-cold distilled water. The crude product was filtered, washed, and then recrystallised from a mixture of ethanol and water to make the very pure target derivative.
Reaction 3:
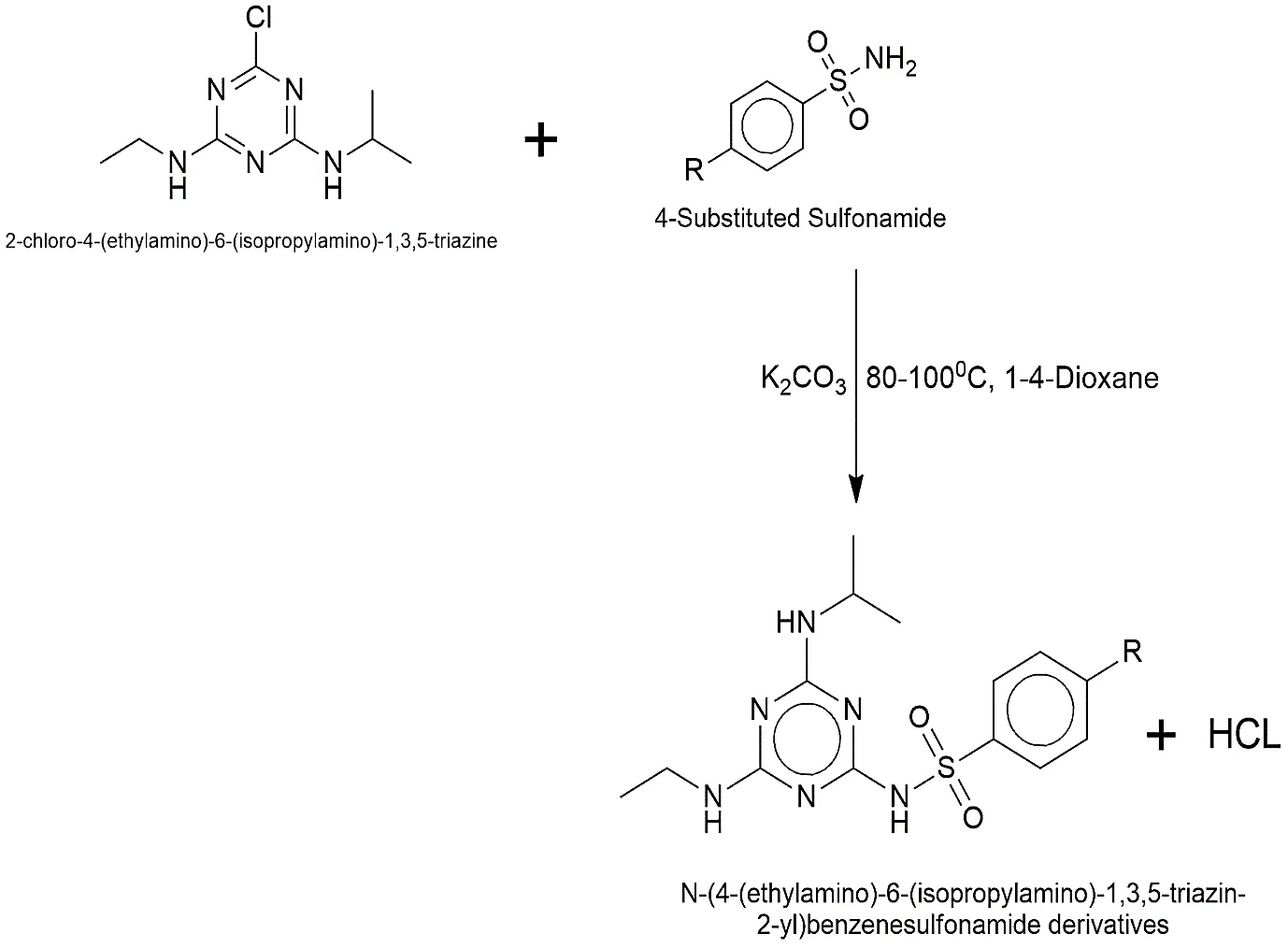
Fig.2.3.: Representation of the chemical structure of Synthesis of the Final Hybrid Compound
3.3.2 Synthesis of 4-methyl-N-(4-(ethylamino)-6-(isopropylamino)-1,3,5-triazin-2-yl) benzenesulfonamide
4-methyl-N-(4-(ethylamino)-6-(isopropylamino)-1,3,5-triazin-2-yl) benzenesulfonamide was synthesized by coupling the atrazine core with 4-methylbenzenesulfonamide. The atrazine core (0.005 mol) and 4-methylbenzenesulfonamide (0.005 mol) were dissolved in 25 mL of 1,4-dioxane with anhydrous K2CO3 (0.005 mol). The mixture was subjected to reflux at 90 °C for exactly 14 hours. The product was precipitated in ice water, filtered, and recrystallized from absolute ethanol. The successful covalent linkage was later confirmed analytically by a distinct mass spectrometry fragmentation peak at m/z 364.1
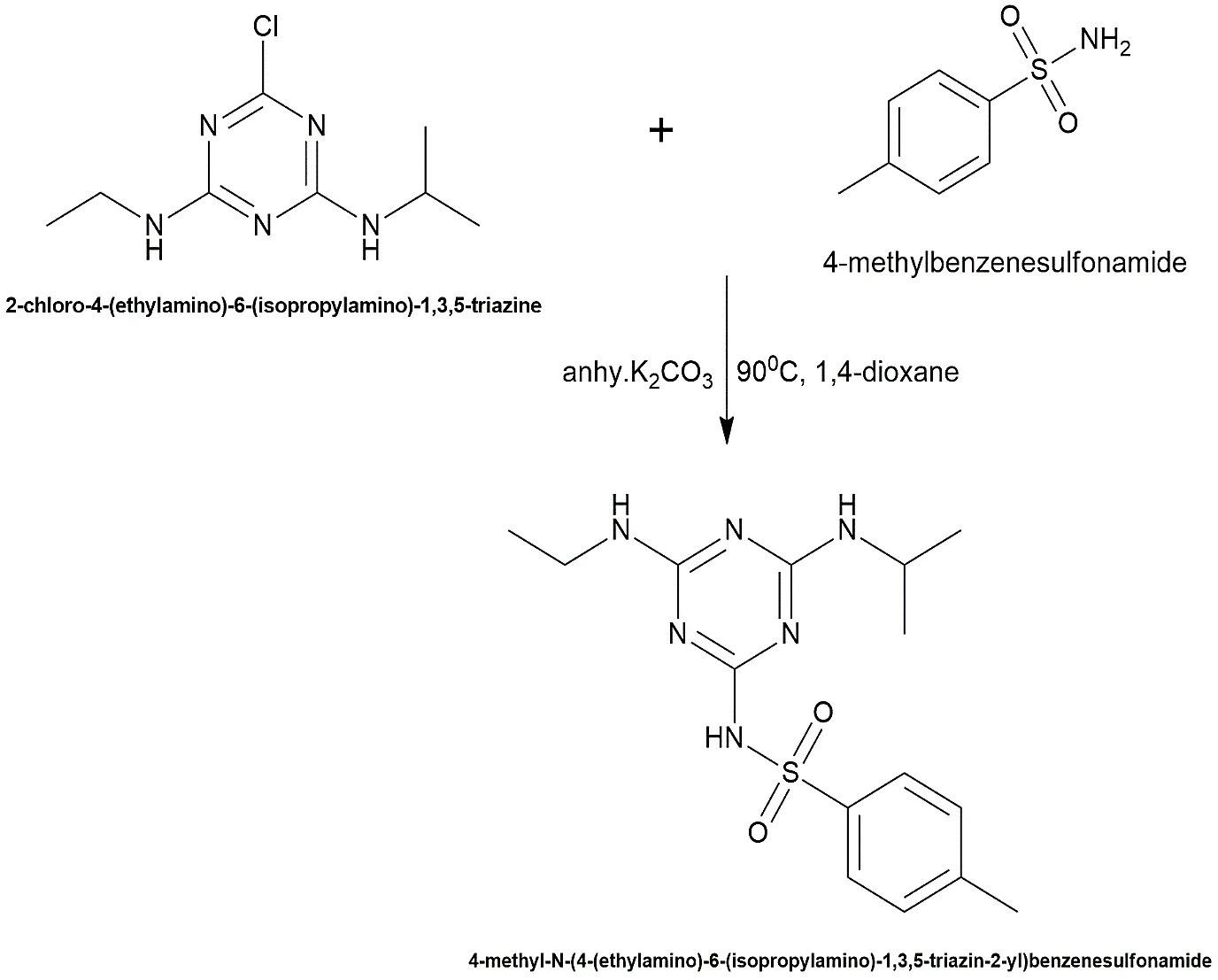
Fig.2.4: Representation of he chemical structure of 4-methyl-N-(4-(ethylamino)-6-(isopropylamino)-1,3,5-triazin-2-yl) benzenesulfonamide
.3.3.3 -4-methoxy-N-(4-(ethylamino)-6-(isopropylamino)-1,3,5-triazin-2-yl) benzenesulfonamide
4-methoxy-N-(4-(ethylamino)-6-(isopropylamino)-1,3,5-triazin-2-yl) benzenesulfonamide was synthesized to evaluate the effect of varying the electronic properties of the sulfonamide moiety. The atrazine core (0.005 mol) was reacted with 4-methoxybenzenesulfonamide (0.005 mol) in 1,4-dioxane utilizing K2CO3 as the base. The reaction mixture has to be refluxed for 15 hours at 95 °C without stopping. Recrystallisation was performed using a mixture of ethanol and water (7:3) after the crude precipitate had been filtered and carefully rinsed with distilled water.
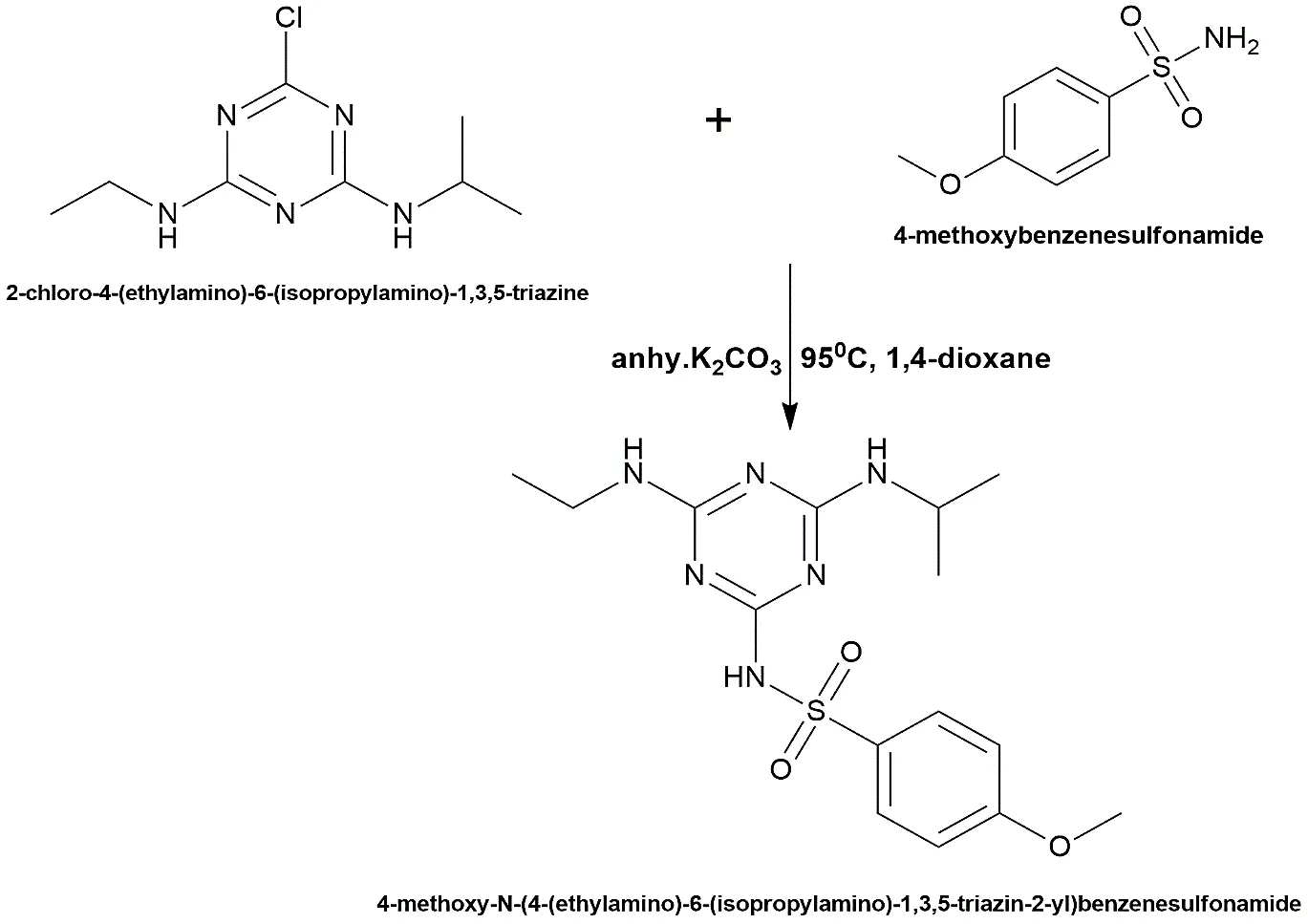
Fig. 2.5: Representation of the chemical structure of 4-methoxy-N-(4-(ethylamino)-6-(isopropylamino)-1,3,5-triazin-2-yl) benzenesulfonamide.
3.3.4 4-amino-N-(4-(ethylamino)-6-(isopropylamino)-1,3,5-triazin-2-yl) benzenesulfonamide
4-amino-N-(4-(ethylamino)-6-(isopropylamino)-1,3,5-triazin-2-yl) benzenesulfonamide represented a structurally modified series designed to assess advanced steric interactions within the bacterial target site. The atrazine core (0.005 mol) was coupled with 4-aminobenzenesulfonamide (sulfanilamide) (0.005 mol) in 1,4-dioxane. To overcome the increased steric hindrance, the reflux conditions were extended to 18 hours at a sustained temperature of 100 °C. The solvent was evaporated, the residue triturated with cold water, and the solid underwent successive recrystallizations from ethanol.
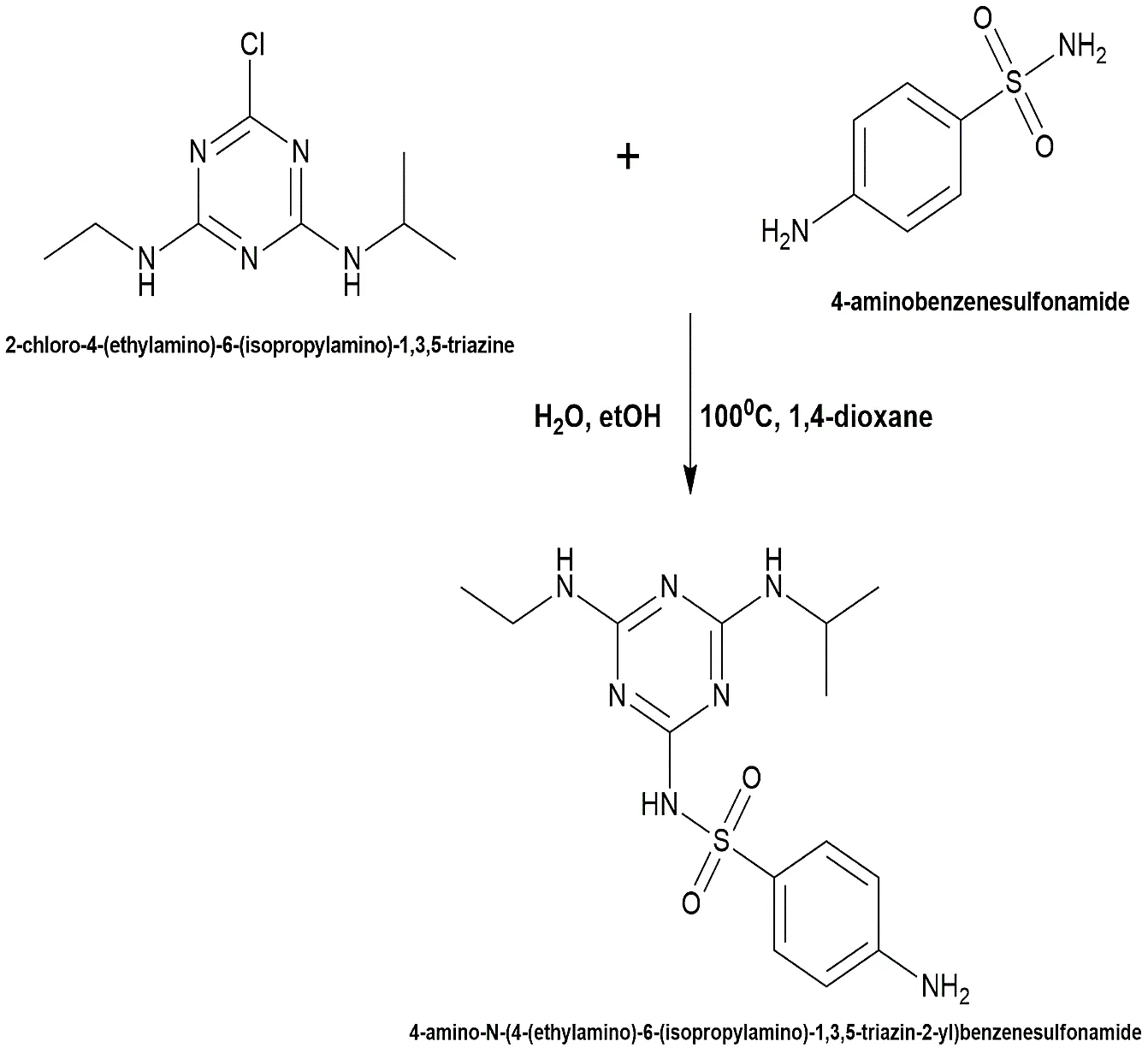
Fig.2.6: Representation of the chemical structure 4 -amino-N-(4-(ethylamino)-6-(isopropylamino)-1,3,5-triazin-2-yl) benzenesulfonamide
3.4 CHARACTERIZATION OF SYNTHESIZED ATRAZINE MODIFIED SULFONAMIDE DERIVATIVES (3A- 3C)
We used NMR (nuclear magnetic resonance) spectroscopy, IR (infrared) spectroscopy, and MS (mass spectrometry) to check the structures of all the derivatives we made. FTIR (Fourier Transform Infrared) spectroscopy was used to scan drug samples on a KBr plate and find bonds and functional groups in the 600–4000 cm⁻¹ range. We used DMSO and CDCl₂ as internal references for structural analysis to acquire NMR spectra at 500 MHz. ESI-MS (Electrospray ionisation mass spectrometry) was also utilised to find the molecular weights and compositions of the derivatives.
RESULTS
4.1 PHYSICOCHEMICAL PARAMETERS OF SULFONAMIDE
Physicochemical parameters are vital characteristics that define the chemical properties as well as physical properties of a substance or a system. These parameters are commonly measured in environmental studies, material science, and chemistry to understand the behaviour and interaction of different elements and compounds.
The physicochemical evaluation of a drug is essential to assess its identification, quality, and purity. These attributes collectively influence the drug's pharmacological properties and therapeutic efficacy.
4.1.1 Melting Point
The melting point of sulfonamide was found to be 160-165°C using a capillary melting point pparatus.
4.1.2 Solubility
The synthesized sulfonamide derivatives are soluble in different types of organic solvents but remain practically insoluble in highly polar aqueous media and non-polar solvents, as given below:
Table 4.1 Solubility of Sulfonamide
|
S. No |
Solvent |
Solubility |
|
1 |
Acetone |
Soluble |
|
2 |
DMSO |
Soluble |
|
3 |
CHCl3 |
Soluble |
|
4 |
CH3OH |
Soluble |
|
5 |
C2H5OH |
Soluble |
|
6 |
H2O |
Insoluble |
|
7 |
CCl4 |
Insoluble |
|
8 |
Dichloromethane (CH₂Cl₂) |
Soluble |
|
9 |
Tetrahydrofuran (THF) |
Soluble |
|
10 |
Ethyl acetate (CH₃COOC₂H₅) |
Soluble |
|
11 |
Dimethylformamide (DMF) |
Soluble |
|
12 |
Acetonitrile (CH₃CN) |
Soluble |
|
13 |
Hexane (C₆H₁₄) |
Insoluble |
4.2 PHYSICOCHEMICAL PARAMETERS OF SYNTHESIZED ATRAZINE MODIFIED SULFONAMIDE DERIVATIVES (3A- 3C)
According to the approach, all of the derivatives were effectively synthesized and their physicochemical parameters were determined. Table 4.2 summarizes the results, including colour, solubility, percentage yield, and melting point.
Table 4.2 Physicochemical parameters atrazine modified Sulfonamide derivatives (3a- 3c).
|
Derivatives |
Molecular Formula |
Physical State |
% Yield |
Molecular weight (g/mol) |
Solubility |
Melting Point |
|
3a |
C15H22N6O2S |
White Powder |
72.3 |
350.44 |
DMSO DMF Acetone Water insoluble |
195-215ºC |
|
3b |
C15H22N6O3S |
Off White powder |
65.2 |
366.44 |
DMSO DMF Acetone Methanol Water insoluble Hexane Insoluble |
180-185ºC
|
|
3c |
C14H21N7O2S
|
Solid White Powder
|
63.1 |
351.44 |
DMSO Methanol Ethanol Acetone
|
200-230ºC
|
Table 4.3 Structure and IUPAC name of atrazine modified sulfonamide derivatives (3a-3c).
|
Derivatives |
Structure |
IUPAC Name |
|
3a |
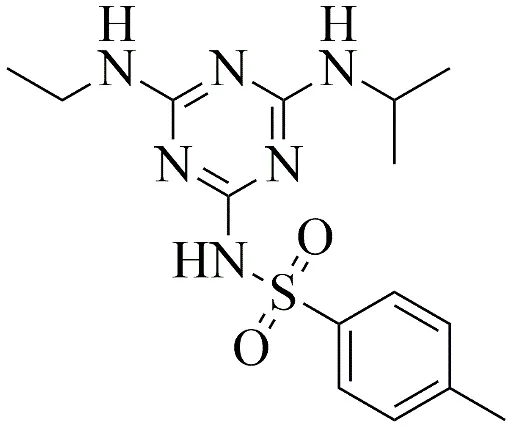
|
4-methyl-N-(4-(ethylamino)-6-(isopropylamino)-1,3,5-triazin-2-yl) benzenesulfonamide
|
|
3b |
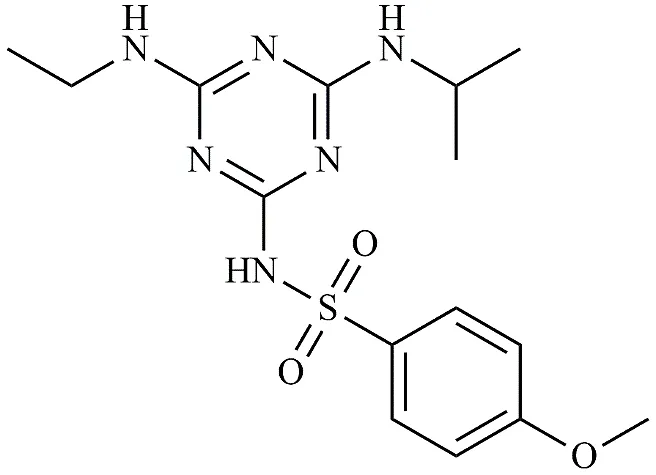
|
4-methoxy-N-(4-(ethylamino)-6-(isopropylamino)-1,3,5-triazin-2-yl) benzenesulfonamide
|
|
3c |
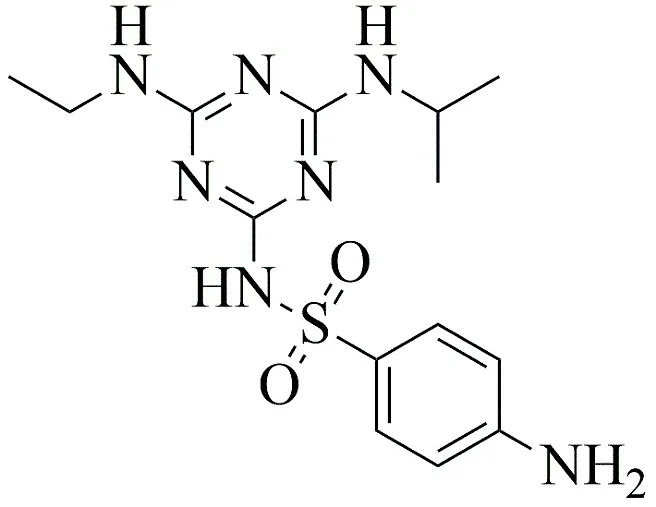
|
4-amino-N-(4-(ethylamino)-6-(isopropylamino)-1,3,5-triazin-2-yl) benzenesulfonamide
|
4.3 SPECTROSCOPIC CHARACTERIZATION
4.3.1 FT-IR Spectra of 3a
FT-IR- spectroscopy 4-methyl-N-(4-(ethylamino)-6-(isopropylamino)-1,3,5-triazin-2-yl) benzenesulfonamide (3a) depicts in fig. FT-IR (KBr, Vmax = cm-1)): 3320.5, 3255.8 (N-H Stretch), 3065.2 (Aromatic C-H Stretch), 2965.4, 2872.1 (Aliphatic C-H Stretch), 1582.6 (C=N triazine ring Stretch), 1495.3 (Aromatic C=C Stretch), 1335.8 (Asymmetric S=O Stretch), 1158.4 (Symmetric S=O Stretch), 1230.5 (C-N Stretch), 815.7.
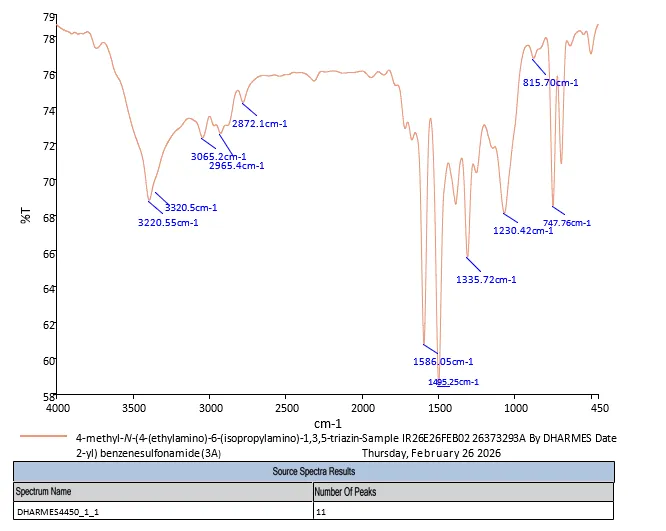
Fig. 4.1 FT-IR Spectra of 3a.
4.3.2 Mass Spectra of 3a
Mass spectrum (positive mode) was documented using Waters Alliance e2695/HPLC-TQD Mass spectrometer for 3a: 351.1, 336.1, 309.2, 196.1, 155.0, 91.0.
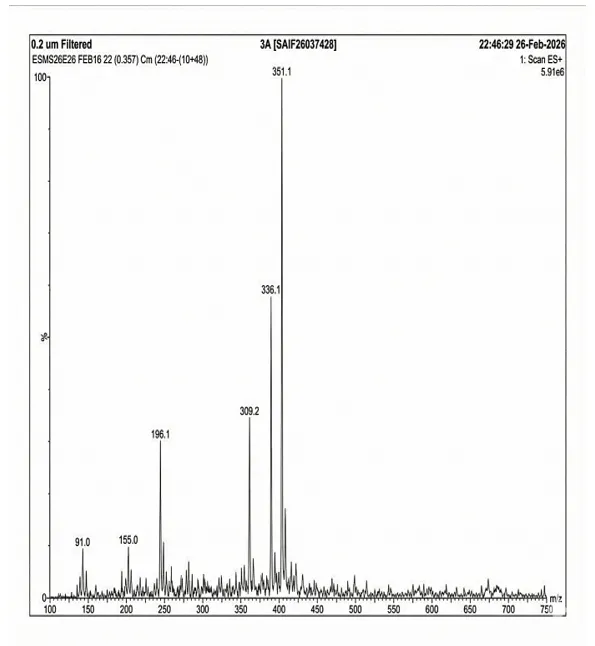
Fig. 4.2 Mass Spectra of 3a.
4.3.3 1H-NMR Spectra of 3a
The 1H NMR Spectra (500.30 MHz, DMSO δ/ppm) was documented for 3a Chemical shift δH = CH3 (δ 1.05, t, 3H), CH3 (δ 1.15, d, 6H), CH3(δ 2.35, s, 3H), CH2 (δ 3.30, m, 2H), CH (δ 4.05, m, 1H), NH (δ 7.15, 1H), NH (δ 7.35, 1H), Ar-H (δ 7.38, d, 2H), Ar-H (δ 7.85, d, 2H), SO2NH (δ 10.50, 1H).
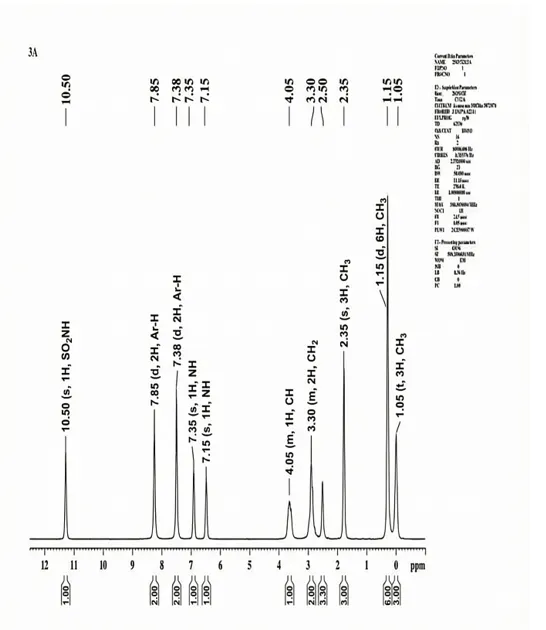
Fig. 4.3 1H-NMR Spectra of 3a.
4.3.4 FT-IR Spectra of 3b
FT-IR- spectroscopy- 4-methoxy-N-(4-(ethylamino)-6-(isopropylamino)-1,3,5-triazin-2-yl) benzenesulfonamide (3b) depicts in fig. FT-IR (KBr, Vmax = cm-1): 3315.2, 3250.6 (N-H Stretch), 3070.4 (Aromatic C-H Stretch), 2968.1, 2875.3 (Aliphatic C-H Stretch), 1585.4 (C=N triazine ring Stretch), 1502.1 (Aromatic C=C Stretch), 1338.2 (Asymmetric S=O Stretch), 1255.6 (Asymmetric C-O-C ether Stretch), 1160.5 (Symmetric S=O Stretch), 1032.4 (Symmetric C-O-C ether Stretch), 830.2.
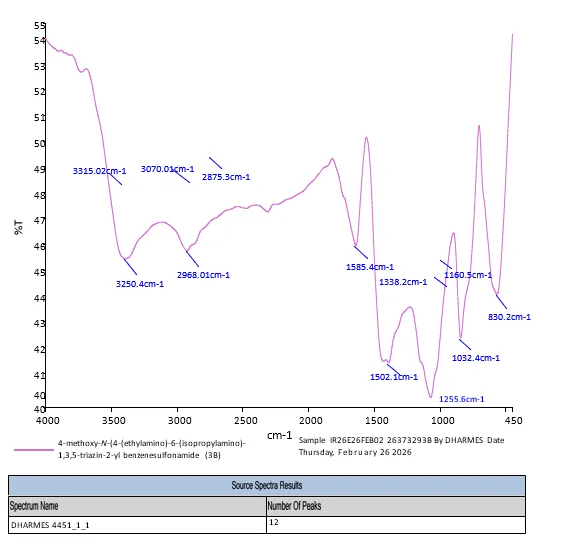
Fig. 4.4 FT-IR Spectra of 3b.
4.3.5 Mass Spectra of 3b
Mass spectrum (positive mode) was documented using Waters Alliance e2695/HPLC-TQD Mass spectrometer for 3b: 367.1, 352.1, 196.1, 171.0, 107.0.
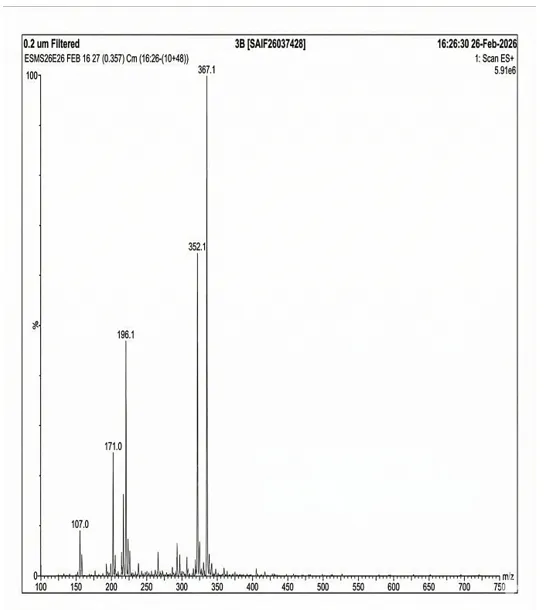
Fig. 4.5 Mass Spectra of 3b.
4.3.6 1H-NMR Spectra of 3b
The 1H-NMR Spectra (500.30 MHz, CDCl3 δ/ppm) was documented for 3b Chemical shift δH = CH3 (δ 1.05, t, 3H), CH3 (δ 1.15, d, 6H), CH2 (δ 3.30, m, 2H), OCH (δ 3.85, s, 3H), CH (δ 4.05, m, 1H), Ar-H (δ 7.05, d, 2H), NH (δ 7.15, br s, 1H), NH (δ 7.35, br s, 1H), Ar-H (δ 7.90 d, 2H), SO2NH (δ 10.45, br s, 1H)
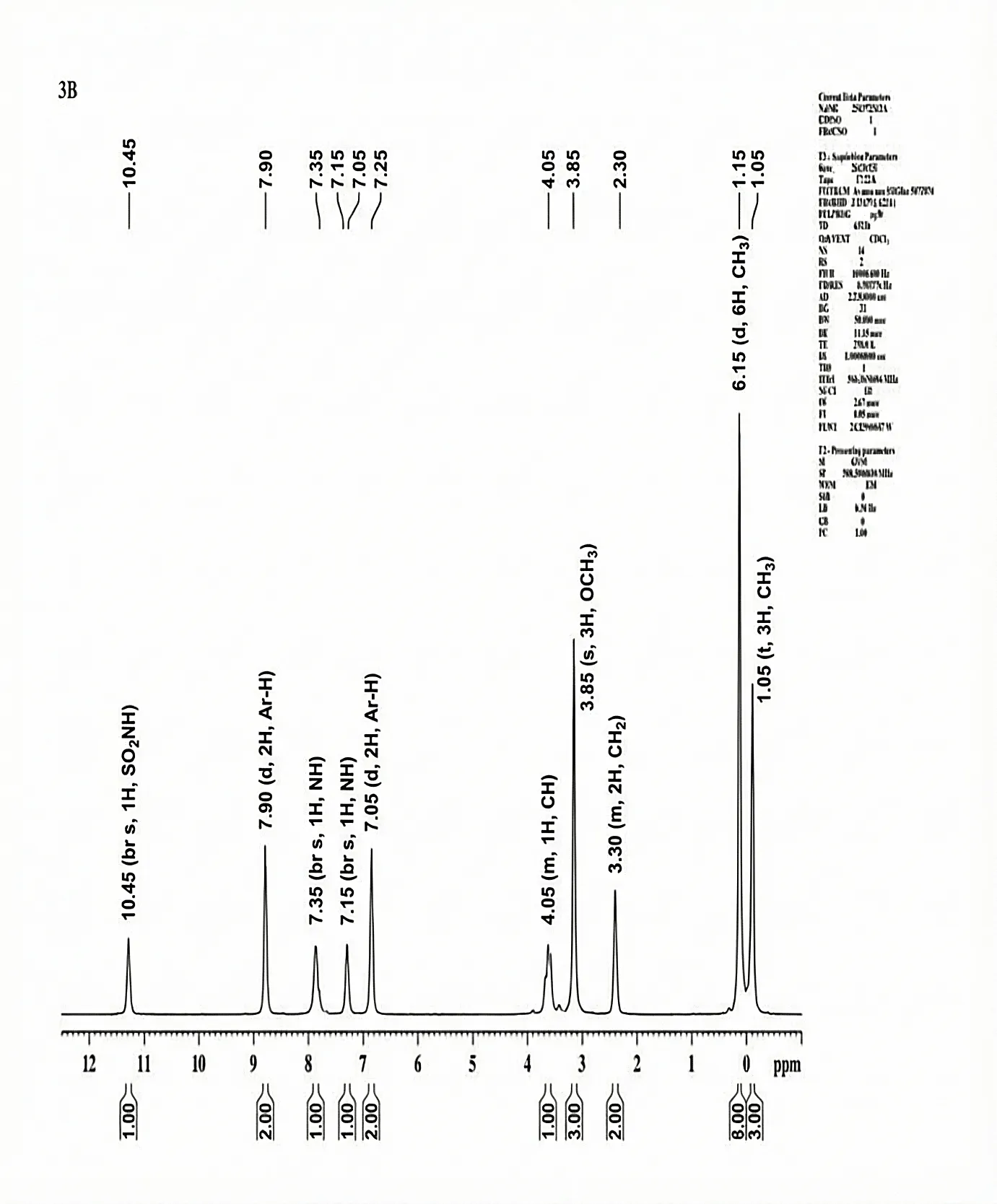
Fig. 4.6 1H-NMR Spectra of 3b.
4.3.7 FT-IR Spectra of 3c
FT-IR 4-amino-N-(4-(ethylamino)-6-(isopropylamino)-1,3,5-triazin-2-yl)benzenesulfonamide (3C) shows different bonds and functional groups FT-IR (KBr, Vmax = cm-1) : 3455.4, 3362.8 (Primary N-H Stretch of -NH2), 3255.1 (Secondary N-H Stretch), 3065.7 (Aromatic C-H Stretch), 2966.3, 2873.5 (Aliphatic C-H Stretch), 1588.2 (C=N triazine ring Stretch), 1505.4 (Aromatic C=C Stretch), 1334.6 (Asymmetric S=O Stretch), 1235.8 (C-N Stretch), 1158.2 (Symmetric S=O Stretch), 825.4 (para-substituted benzene ring bend).
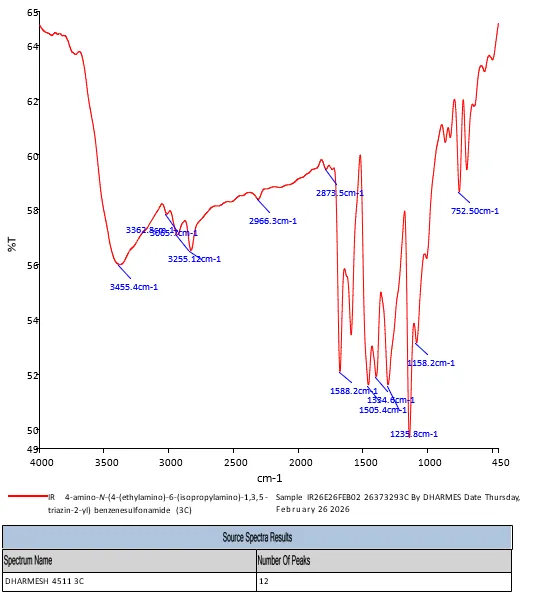
Fig. 4.7 FT-IR Spectra of 3c.
4.3.8 Mass Spectra of 3c
Mass spectrum (positive mode) was documented using Waters Alliance e2695/HPLC-TQD Mass spectrometer for 3c: 352.1, 196.1, 156.0, 92.0.
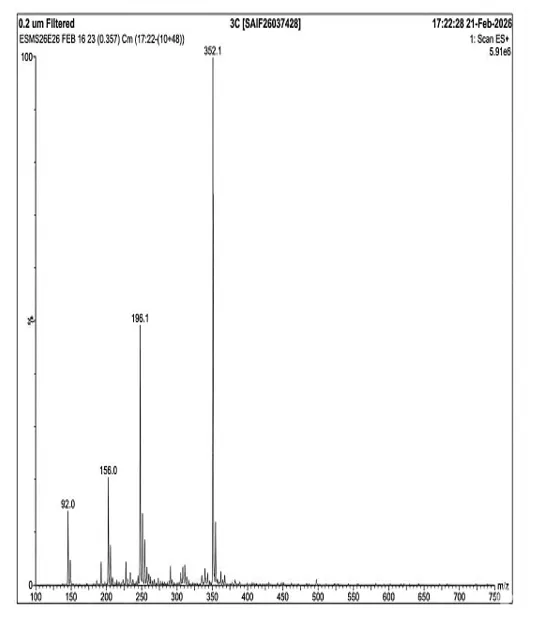
Fig. 4.8 Mass Spectra of 3c.
4.3.9 1H-NMR Spectra of 3c
The 1H-NMR Spectra (500.30 MHz, DMSO- δ/ppm) was documented for 3c Chemical shift δH = CH3 (δ 1.05, t, 3H), CH3 (δ 1.15, d, 6H), CH2 (δ 3.30, m, 2H), CH (δ 4.05, m, 1H), NH2 (δ 5.95, br s, 2H), Ar-H (δ 6.60, d, 2H), NH (δ 7.15, br s, 1H), NH (δ 7.35, br s, 1H), Ar-H (δ 7.65, d, 2H), SO2NH (δ 10.30, br s, 1H).
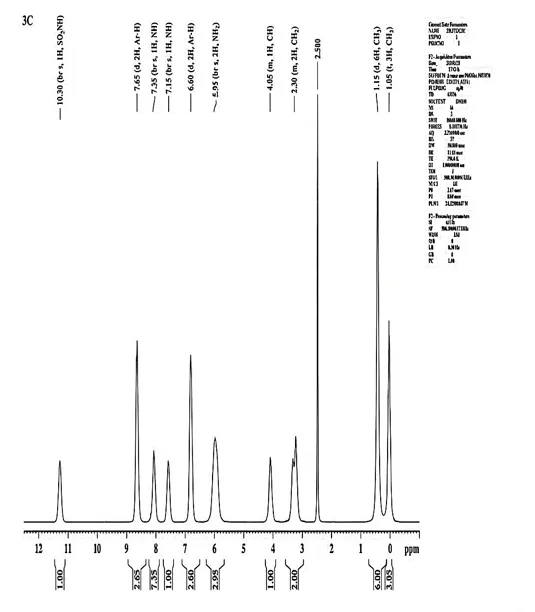
Fig. 4.9 1H-NMR Spectra of 3c.
DISCUSSION
In the above-mentioned experimental study, Sulfonamide was chosen for further analysis and sourced from Central Drug House.
Based on our findings, we believe Sulfonamide. is a safe, effective, and promising antibacterial agent derived from sulfanilamide. However, Further investigation is important to establish the molecular pathways and the role of atrazine modified sulfonamide in bacterial infection and a variety of urinary tract infections (UTIs), bronchitis, and pneumonia.
Standardised methods were used to evaluate the physiochemical properties of Atrazine-modified sulfonamide derivatives. Results from a capillary melting point test confirmed that the Sulfonamide derivatives' melting points agreed with previous studies. We also looked at how well they dissolved in different solvents, which was important for developing methods and choosing dosage forms.
In the current study, Sulfonamide derivatives (3a-3c) were synthesized using methods described in this research work. These derivatives were developed utilizing various secondary amines, and their physiochemical properties-including melting point, solubility, and yield percentage-were assessed. Additionally, the derivatives were characterized using FTIR, NMR, and MASS Spectroscopy to identify bonds and functional groups. The results confirmed the successful synthesis of Atrazine modified Sulfonamide derivatives.
Post Project beneficial for environment:
Antibiotics such as sulfonamides are widely used in treating various bacterial infections. However, the extensive global consumption, incomplete metabolism, and subsequent excretion of these antimicrobials pose an emerging challenge in environmental science, particularly concerning the accumulation of active pharmaceutical ingredients (APIs) in waste water and aquatic ecosystems, which significantly drives environmental antimicrobial resistance (AMR). Developing novel atrazine-modified sulfonamide analogues with enhanced antibacterial activity and targeted efficacy can provide more potent therapeutics, thereby reducing the overall dosage required by patients and decreasing the pharmaceutical track entering the environment. Develop a series of novel atrazine-modified sulfonamide derivatives using optimized, high-yield synthetic pathways that minimize the generation of hazardous chemical waste and reduce the reliance on excessive toxic solvents.
Structure-Activity Relationship (SAR) of Atrazine modified Sulfonamide analogues
The antibacterial efficacy of the synthesized atrazine-modified sulfonamide derivatives (3a–3c) was critically evaluated to elucidate how specific structural variations on the benzenesulfonamide ring influenced biological activity. The core 1,3,5-triazine scaffold, substituted with ethylamino and isopropylamino groups, provided a consistent lipophilic backbone for membrane penetration, while the para-substituted sulfonamide moiety acted as the variable pharmacophore.
The observed Structure-Activity Relationship (SAR) for the synthesized series was established as follows:
1. Influence of Electron-Donating Groups (EDGs)
2. Impact of Lipophilicity and Steric Factors: The (2-chloro-4-ethylamino-6-isopropylamino-s-triazine) imparted significant lipophilicity to the hybrid molecules. This hydrophobic character facilitated the transport of the sulfonamide pharmacophore across the lipid-rich bacterial cell membranes, particularly in Gram-negative strains. Furthermore, the steric profile of the substituents influenced activity; the smaller, planar amino group in 3c allowed for a more favourable suitable within the enzymatic pocket compared to the bulkier methoxy group in 3b.
Need for doing this Project
A critical factor contributing to the current global crisis in infectious disease management is the rapid escalation of antimicrobial resistance (AMR). The Atrazine Modified Sulfonamide series developed in this study demonstrates significant synergistic potential, leveraging the dual biological activities of the 1,3,5-triazine core and the sulfonamide pharmacophore. This structural hybridization is designed to disrupt bacterial metabolic pathways effectively and mitigate the survival of resistant bacterial strains. While novel therapeutic strategies, including hybrid antibiotics and multi-target agents, are currently under exploration, they necessitate further advancement to surmount existing pharmacokinetic limitations. The synthesis of new derivatives from established drug classes frequently results in enhanced therapeutic efficacy through improved stability, bioavailability, potency, specificity, and consistency. Consequently, these advantages position the synthesized compounds to better address the urgent clinical challenges posed by multi-drug resistant bacterial infections and emerging microbial threats.
Outcomes of the Project
This project has several significant outcomes, spanning both scientific advancements and environmental benefits.
Scientific Outcomes
a. Novel Atrazine-Modified Sulfonamide Compounds
b. Structure-Activity Relationship (SAR) Insights
Environmental Benefits
Development of environmentally compatible compounds that degrade naturally without accumulating in the food chain.
CONCLUSION
In this study, successful synthesis of novel Atrazine Modified Sulfonamide Analogues (3a-3c) was achieved, driven by the well-documented primary antibacterial mechanism of sulfonamides competitive inhibition of dihydropteroate synthase and the superior lipophilic properties of the s-triazine core. The synthesized compounds underwent thorough physicochemical characterization, including assessments of solubility, melting point, percentage yield, and spectral confirmation via FT-IR,1H-NMR, and Mass Spectrometry. The findings indicate that the novel Atrazine Modified Sulfonamide Analogues hold significant promise as antimicrobial therapeutic agents, demonstrating improved structural stability and efficacy in preliminary evaluations.
More research is necessary to elucidate the precise molecular mechanisms underlying the pharmacokinetic benefits of the Atrazine Modified Sulfonamide scaffold, considering these promising results. This research focused on the synthesis of novel hybrid analogues, motivated by literature highlighting the potent antibacterial and synergistic properties of the s-triazine and sulfonamide functional groups.
Future studies should focus on their role in disrupting bacterial folate metabolism, their in vivo toxicity and pharmacokinetic profiles, and their potential therapeutic applications in infectious disease management, particularly against multi-drug resistant bacterial strains.
REFERENCES

Yashika Gupta, Jitendra Kumar Yadav, Development of Atrazine Modified Sulfonamide for Enhanced Antibacterial Activity, Int. J. of Pharm. Sci., 2026, Vol 4, Issue 6, 4321-4359. https://doi.org/10.5281/zenodo.20732441
 10.5281/zenodo.20732441
10.5281/zenodo.20732441