
We use cookies to ensure our website works properly and to personalise your experience. Cookies policy
College Of Pharmacy,Madras Medical College, Chennai-03
Tuberculosis remains one of the major global threats to human population due to complications such as presence of multi drug resistant strains of mycobacterium tuberculosis, toxicity and poor tolerability of traditional first line anti-tubercular drugs [1,2]. Oxazolidinone are five membered heterocyclic compounds belonging to class of antibiotics that exert their activity by inhibiting bacterial protein synthesis. In this study, a docking guided MD simulation approach was studied for novel oxazolidin-2-one derivatives as anti-tubercular agents targeting peptidyl transferase centre of 50s ribosomal subunit [3,4]. Molecular docking was performed to evaluate binding affinity and interaction patterns of novel derivatives against the protein PDB ID - 8IPK [5]. After which md simulation studies was performed to evaluate protein ligand stability. In addition to this, lead optimization was performed tools like Swiss ADME, ADMET lab, pass prediction, Osiris property explorer to obtain derivatives with favourable pharmacokinetic or ADME profile. These optimized derivatives showed strong ribosomal binding, preserved compactness of protein structure and greater inhibitory potential against mycobacterium tuberculosis. Through these findings, we can tell that oxazolidin-2-one derivatives are said to promising lead candidates for tuberculosis treatment
Tuberculosis is still said to be a disease of concern even after availability of traditional first line drugs such as isoniazid and pyrazinamide [1,6]. Inhibiting protein synthesis is a well validated target as it selectively and specifically targets only mycobacterial ribosome instead of affecting human ribosomes and overcomes resistance.
Oxazolidinones are the novel basic compounds which bind to PTC and thereby prevent the formation of functional initiation complex essential for protein synthesis [3,7]. Linezolid is a potent oxazolidinone antibiotic effective against various serious bacterial infections and a great choice for multi drug resistant strains. Hence WHO recommended the use of LZD in 2022 in BPaLM dose regimen. But still, linezolid is not so effective because it causes myelosuppression, optic neuropathy and anaemia [4,8] in patients. Thus, there is a need for discovery of newer potent drugs eliminating the disadvantages of linezolid. Other recent drugs for tuberculosis treatment includes – Bedaquiline, a diarylquinoline approved in 2012 acts by inhibiting ATP synthase enzyme and Delamanid, a nitro imidazooxazole derivative acts by inhibiting mycolic acid synthesis. Due to advancements in computational drug design, discovery of novel lead compounds is become easier. Virtual screening of large number of compounds can be cost efficiently performed before experimental studies [9] which reduce time for the researchers. Molecular docking was performed out of which the most promising lead candidates were further estimated using MD simulation to analyse protein-ligand binding.
2.MATERIALS AND METHODS:
A) PROTEIN AND TARGET SELECTION:
The peptidyl transferase centre of 50s ribosomal subunit of mycobacterium tuberculosis was selected as a validated target due to major involvement in the process of protein synthesis [10,11]. Selection of an PDB structure from protein data bank is a important step for rational drug design. Based on various criteria, PDB ID 8IPK was selected as protein. The 3D high resolution crystallographic structure was obtained from RCSB Protein Data Bank. Protein was prepared by removing excess water molecules, adding hydrogen bonds and applying appropriate charges [5,12].

Fig no.1 3D structure of protein (PDB ID 8IPK)
B) LIGAND PREPARATION:
A library consisting of nearly 100 new lead molecules as potent protein synthesis inhibitors was designed and prepared based on necessary pharmacophoric features such as hydrogen bond acceptor, aromatic ring, etc essential for antibacterial activity [13]. These structures were then evaluated using PubChem to check their novelty. Further, they were energy minimized using Chem3D SOFTWARE and converted to suitable file formats to carry out the docking procedure [14].
C) MOLECULAR DOCKING:
Molecular docking was performed for various novel derivatives with the help of AUTODOCK Vina software. Binding affinity, binding pose and binding interactions was calculated against the protein [15]. Compounds with more negative binding energy and stable affinity were identified as high promising candidates.
D) MD SIMULATION STUDIES:
After performing molecular docking, the drug candidates with highly stable binding energy were further evaluated for its protein-ligand stability by MD simulations. GROMACS and DESMOND software were used to evaluate RMSD, RMSF, Radius of gyration and torsion profile of ligand [16]. This provided us information about the stability of protein and ligand throughout the simulation studies and correlation with anti-tubercular activity.
E) ADME AND TOXICITY PREDICTION:
ADME prediction of the designed ligands or molecules is done by using tools like Swiss ADME [17] and ADMET Lab 2.0 [18]. This software evaluated properties such as lipophilicity, BBB penetration, oral bioavailability and major rules like Lipinski’s rule of five. Pass prediction is a software used to predict the general biological potential of drug like candidates against various biological targets. Toxicity risks such as carcinogenicity, mutagenicity, reproductive toxicity and irritation were evaluated using OSIRIS Property Explorer [19].
3. RESULTS AND DISCUSSION:
3.1 MOLECULAR DOCKING ANALYSIS:
Molecular docking is generally used to predict the protein-ligand interaction and orientation at specific binding pocket determined using AUTO Grid software [15]. Several novel derivatives were found with highly stable binding affinity and energy, out of which two ligands CDD14 and CDD53 were found with highest promising anti-tubercular activity [20]. Table 1 presents the numerical values of binding energy and interactions of the two best ligands. Software involved-AUTO DOCK (1.5.7) tool [21].
TABLE 1 represents numerical values of binding energy and amino acid interaction of ligands.
|
SNO
|
COMPOUIND |
BINDING ENERGY (kcal/mol) |
AMINO ACID INTERACTIONS |
|
1
|
CDD14

|
-10.3 |
HIS A:210 ARG A:207 LEU A:117 PHE A:115 PHE A:216 ARG A:220 CYS A:103 |
|
2 |
CDD53

|
-8.83 |
CYS A:103 GLY A:32 SER A:30 SER A:30 HIS A:33 LEU A: 98 |
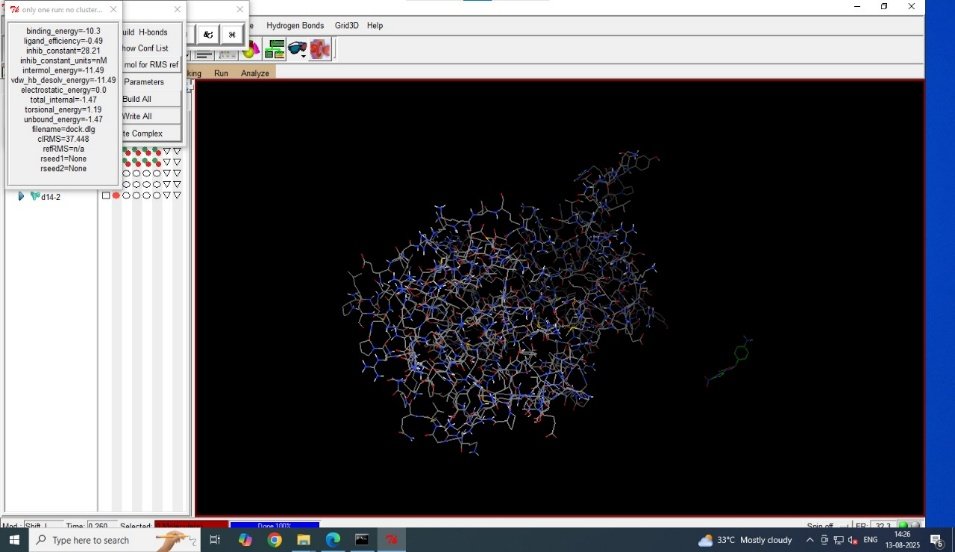
Fig no.2a CDD14 molecular docking with binding energy
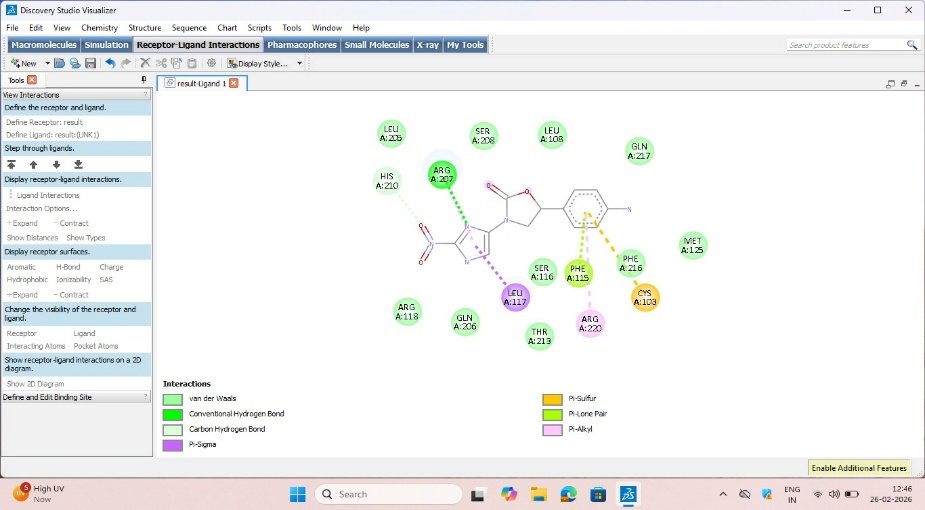
Fig no.2b CDD14 binding interactions

Fig no.3a CDD53 molecular docking with binding energy

Fig no.3b CDD53 binding interactions
3.2 MOLECULAR DYNAMICS SIMULATION:
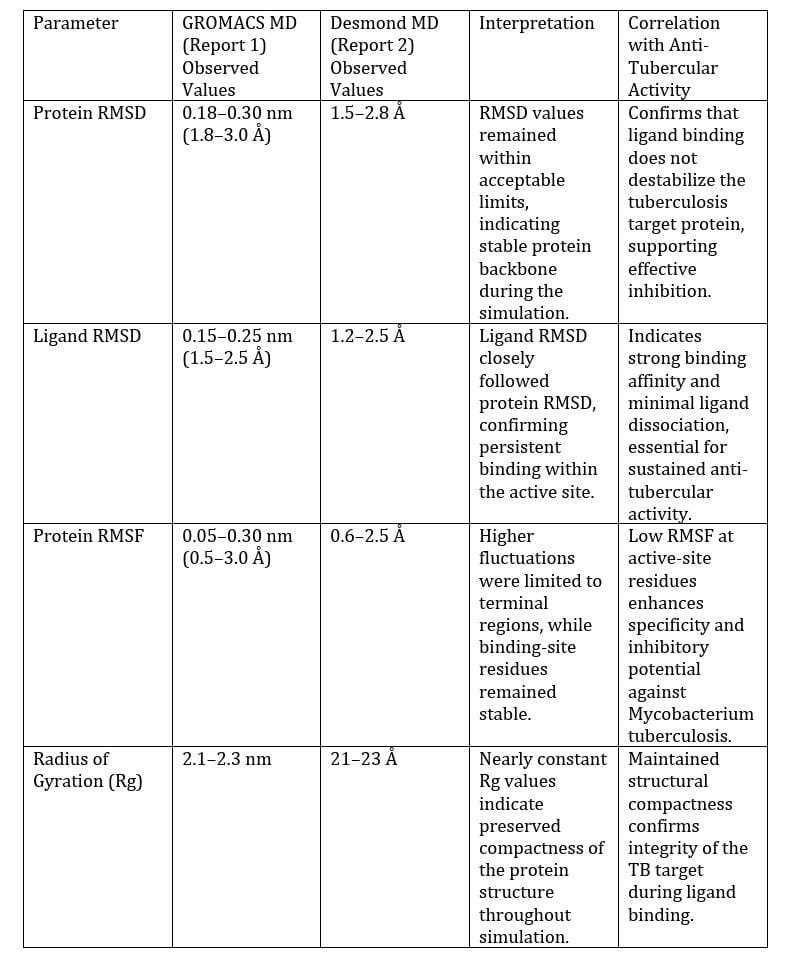
The two best ligands were further studied for their protein-ligand stability by observing the motion of atoms and molecules over time based on the concept of newtons law of motion defined by various force fields. The process of md simulation involves- system preparation, energy minimization, heating and thermalization, equilibration and MD run. Both software GROMACS [22] and DESMOND [23] was used for the process with necessary system requirements such as temperature-300K and simulation time-100.102ns. The various properties analysed are Root-Mean-Square Deviation (RMSD)- for structural stability, Root-Mean-Square Fluctuation (RMSF)- for per-residue flexibility, Hydrogen bond and distance analysis – talks about interaction dynamics and then Energy components [16].

Fig no.4 CDD14 MD simulation using GROMACS a) RMSD b) RMSF c) Radius of gyration

Fig no.5 CDD53 MD simulations using DESMOND a) RMSD b) RMSF c) protein secondary structure
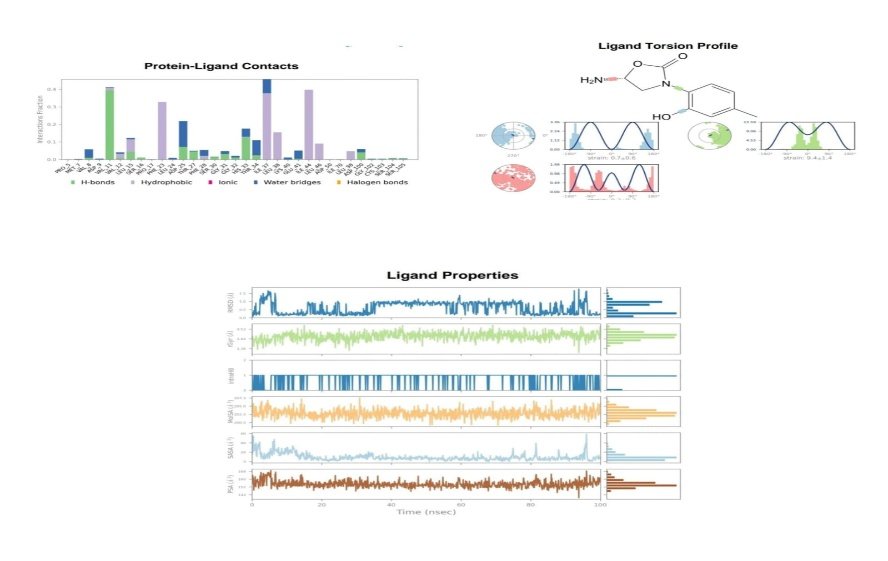
Fig no.6 CDD53 MD simulations using DESMOND a) protein-ligand contacts b) ligand torsion profile c) ligand properties
TABLE 2 presents comparison between two MD simulation software GROMACS and DESMOND for CDD14 and CDD53 molecules respectively along with its correlation to anti tubercular activity.
3.3 ADME AND TOXICITY PREDICTION:
With the help of ADMET tools such as SWISS ADME [17], ADMET Lab [18], OSIRIS property explorer [19] novel derivatives with favourable pharmacokinetic profiles were obtained. Favourable pharmacokinetic profile ensures better bioavailability, lesser side effects and improved tolerability of the drug like candidates. Pass prediction provides data about all possible potential of a molecule for various biological targets.
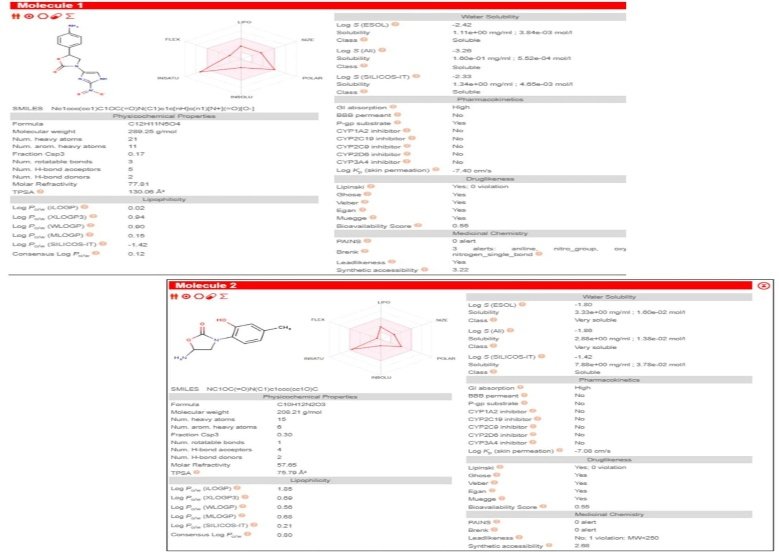
Fig no.7 SWISS ADME prediction a) CDD14 b) CDD53
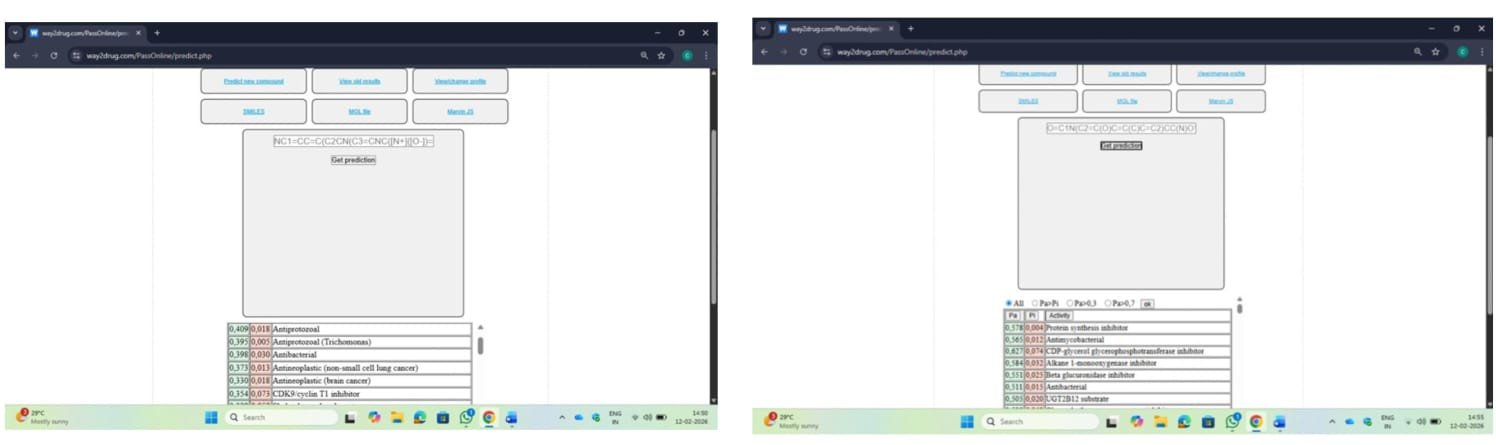
Fig no.8 PASS PREDICTION a) CDD14 b) CDD53

Fig no.9 OSIRIS Property Explorer a) CDD14 b) CDD53
CONCLUSION
As discussed earlier, due to longer duration of traditional treatment and higher multi drug resistant strains of mycobacterium tuberculosis, the need for newer standard anti-TB drugs is increased. The above study provides two major candidates CDD14 and CDD53 with binding energy -10.3 and -8.86 kcal/mol respectively. These candidates demonstrated stronger ribosomal binding, limited fluctuations in protein structure, stable dynamic behaviour and favourable pharmacokinetic profile. Results from MD simulations further made stronger note on protein-ligand stability. Therefore, this indicates oxazolidine-2-one derivatives have greater potential in the treatment of anti-tuberculosis therapy. Invitro and experimental studies can further validate the study [9,10,11].
REFERENCES




M. Sathish, Chethna Jain C., Deepika S., Dharshini G., Docking Guided Md Simulation Study of Novel Oxazolidin-2-One Derivatives as Anti-Tubercular Activity Targeting 50s Ribosomal Subunit, Int. J. of Pharm. Sci., 2026, Vol 4, Issue 3, 2491-2499. https://doi.org/10.5281/zenodo.19147311
 10.5281/zenodo.19147311
10.5281/zenodo.19147311