
We use cookies to ensure our website works properly and to personalise your experience. Cookies policy
Srinath college of pharmacy, chh. Sambhajinagar.
Solid oral dosage forms remain the foundation of modern therapeutics due to their patient convenience, stability, and cost-effective manufacturing. This review offers a comprehensive look into the evaluation tests for both tablets and capsules, connecting established pharmacopeial requirements from sources like the USP, BP, and IP with practical laboratory methods and recent technological advancements1–3. For tablets, the evaluation process begins with pre-compression tests, such as flow and density indices, and extends to a full range of post-compression quality checks, including weight variation, hardness, friability, disintegration, and dissolution. Conversely, the evaluation of capsules addresses unique shell-related properties like moisture sensitivity, brittleness, and cross-linking, alongside core tests for weight variation, content uniformity, and dissolution.The review includes comparative tables that summarize the key differences in evaluation protocols for tablets and capsules, and it presents a logical workflow diagram that outlines a sensible testing sequence from initial appearance checks to advanced, non-destructive techniques like NIR and Raman spectroscopy. The paper also highlights common challenges, such as capping in tablets and leakage in softgels, and offers best-practice recommendations for immediate-release, modified-release, and special dosage formats. In conclusion, robust evaluation is critical for ensuring dose accuracy, consistent drug release, and patient safety, all of which are vital for regulatory compliance and efficient scale-up of pharmaceutical products
The Enduring Importance of Solid Oral Dosage Forms
Solid oral dosage forms, such as tablets and capsules, are the most prevalent and widely utilized drug delivery systems in the global pharmaceutical market. They constitute over 70% of all formulations dispensed, a testament to their widespread acceptance and utility4–6. Their popularity is not accidental; these forms offer unparalleled convenience for patients, are cost-effective to produce, easy to administer, and simple to transport. A significant advantage of tablets is their versatility, which allows formulators to create a variety of release profiles, including immediate, delayed, and sustained-release versions. Capsules, on the other hand, excel at masking the unpleasant taste of an active pharmaceutical ingredient (API)7,8, protecting sensitive drugs from environmental factors like light and moisture, and delivering both powders and liquids with high dose precision. The development and rigorous evaluation of these dosage forms are foundational to modern pharmaceutical science, ensuring they are not only effective but also safe for patient consumption
A Historical Perspective and Market Dominance
The history of solid oral dosage forms dates back centuries, with ancient civilizations using compressed herbal powders. However, the modern tablet, as we know it, emerged in the mid-19th century with the invention of the tablet compression machine. This innovation revolutionized medicine by standardizing drug delivery and enabling mass production. Similarly, the development of gelatin capsules provided a more palatable alternative for bitter or foul-tasting medicines8,9. These inventions paved the way for the pharmaceutical industry to grow from an apothecary-based model to a large-scale, industrial one.Today, tablets and capsules dominate the market due to their unique balance of efficacy, safety, and manufacturing efficiency4,5. They are the go-to choice for a vast range of therapeutic areas, from over-the-counter pain relievers to life-saving prescription medications. Their market share continues to grow, driven by patient preference and continuous advancements in formulation science. This dominance, however, places a critical emphasis on robust quality control and evaluation to uphold public health standards.
Tablets vs. Capsules: A Comparative Overview
Although they are both solid oral dosage forms, tablets and capsules have distinct
A tablet is a solid preparation containing one or more APIs, which are compacted with excipients into a specific shape. The manufacturing process involves pre-compression evaluation of the powder blend for properties like
flowability and compressibility, followed by post-compression tests on the finished product. Key post-compression tests for tablets include
hardness (crushing strength) and friability, which measure the tablet's resistance to breaking or chipping.
A capsule, conversely, is a unit-dose form where the drug is enclosed within a shell, typically made of gelatin or hydroxypropyl methylcellulose (HPMC). For capsules, evaluation focuses on the unique challenges posed by the shell and its contents. Shell-related issues such as moisture sensitivity, brittleness, and cross-linking are critical considerations16. Consequently, specific tests like moisture permeation and mechanical strength are vital for ensuring capsule integrity.
Despite these differences, tablets and capsules share several core evaluation tests, including
weight variation, content uniformity, disintegration, and dissolution. These shared tests are crucial for verifying that each dosage unit delivers the correct amount of drug at the proper rate.
The Critical Need for Evaluation and Quality Control
The evaluation of solid oral dosage forms is a scientific imperative, not merely a regulatory formality. A failure to perform these tests can lead to severe clinical outcomes, such as under-dosing, which can result in therapeutic failure, or over-dosing, which may cause adverse effects. For instance, a tablet with poor compression might suffer from
capping or lamination, where the top or bottom of the tablet separates from the main body. Similarly, improper storage can lead to
brittleness or cross-linking in capsule shells, affecting their stability and drug release10.
Systematic quality control ensures that every tablet and capsule is manufactured to a consistent standard, ensuring
dose accuracy, predictable drug performance, and patient safety. These tests are also instrumental in streamlining the scale-up process from a laboratory setting to commercial production, minimizing waste and preventing costly regulatory delays.
Regulatory Requirements and Modern Advances
The standards for evaluating tablets and capsules are globally harmonized by official pharmacopeias, including the
United States Pharmacopeia (USP), British Pharmacopoeia (BP), and Indian Pharmacopoeia (IP). These pharmacopeias provide detailed test procedures, acceptable limits, and criteria for various dosage forms. Additionally, international guidelines from the
International Council for Harmonisation (ICH), such as ICH Q6A, Q8, and Q10, provide a framework for a scientifically justified and risk-based approach to quality control11–14.
While traditional tests remain the foundation of quality control, modern technology is revolutionizing the field.
Process Analytical Technology (PAT), Near-Infrared (NIR) spectroscopy, and Raman spectroscopy enable real-time, non-destructive monitoring of critical quality attributes during the manufacturing process10,15. This shift from end-product testing to in-process control enhances efficiency and ensures a higher level of product quality.
The concept of in-vitro/in-vivo correlation (IVIVC) is also advancing, allowing formulators to predict a drug's absorption in the body based on in-vitro dissolution data, thereby reducing the need for extensive clinical trials. This review aims to consolidate and compare these various evaluation methods, providing a comprehensive guide for students, formulators, and researchers16.
This review was compiled through a comprehensive and systematic literature search to ensure the inclusion of all critical evaluation parameters for solid oral dosage forms (tablets and capsules).
Relevant information was meticulously gathered from a variety of authoritative sources:
British Pharmacopoeia (BP) , and the
Indian Pharmacopoeia (IP).
The search used specific keywords to ensure a thorough collection of relevant data. These keywords included "tablet evaluation tests," "capsule evaluation tests," "pre-compression parameters," "post-compression quality control," "dissolution testing," "disintegration studies," "friability," "weight variation," "content uniformity," "stability testing," and "bioavailability".
2.2 Inclusion and Exclusion Criteria
To maintain the quality and focus of the review, specific criteria were applied during the selection of articles and guidelines.
The collected information was systematically classified to provide a structured and comprehensive overview. The data was organized into the following key sections:
To enhance clarity, tables, figures, and flow diagrams were incorporated to visually represent the evaluation procedures. Each section of the review is supported by references to official pharmacopeial standards and relevant scientific studies.
The primary aim of this review is to consolidate and compare the various evaluation tests for tablets and capsules. By highlighting both their similarities and differences, while also integrating modern advancements, this review seeks to provide a valuable resource for researchers, formulators, and students in the field. It is designed to foster a deeper understanding of the scientific and regulatory aspects of quality control for solid oral dosage forms.
3. Evaluation Tests for Tablets
Tablets are the most widely used solid oral dosage form due to their ease of administration, accurate dosing, stability, and cost-effectiveness. However, to guarantee their quality, safety, and efficacy, tablets must undergo a series of systematic evaluation tests both during the formulation phase and after compression. These tests can be broadly categorized into several key areas: pre-compression parameters, post-compression parameters, in-vitro performance tests, and advanced characterization.
3.1 Pre-Compression Parameters
These tests are crucial for assessing the characteristics of the powder blend or granules before they are compressed into a tablet. Poor flow properties in the powder can lead to significant problems during manufacturing, such as weight variation, inconsistent hardness, or sticking to the tablet press, which ultimately compromises the quality of the final product.
The angle of repose measures the flowability of a powder. It is defined as the maximum angle formed between the surface of a pile of granules or powder and the horizontal plane. This angle is determined by allowing the powder to flow through a funnel onto a flat surface, and it is calculated from the height.
(h) and radius (r) of the resulting cone. A lower angle of repose indicates better flow properties, while a higher angle suggests poor flow4,6.
3.1.2 Bulk Density and Tapped Density
These tests provide insights into the packing and compressibility of the powder.
Bulk density (BD) is the mass of the powder divided by its bulk volume, measured by simply pouring the powder into a graduated cylinder.
Tapped density (TD) is determined after mechanically tapping the container until the powder volume no longer changes. These values are used to calculate the compressibility indices of the powder4,6.
3.1.3 Carr's Index (Compressibility Index)
Carr's Index is a measure of a powder's flowability and compressibility, derived from its bulk and tapped densities. It's calculated using the formula: CI = 100 × (ρT - ρB) / ρT.
. A lower index indicates better flow characteristics, with values below 10% considered excellent3,4.
Similar to Carr's Index, Hausner's Ratio is another indicator of a powder's flowability. It is calculated as the ratio of tapped density to bulk density (HR=TD\BD)3,4.
A value closer to 1.0 indicates excellent flow, while values above 1.25 suggest poor flow.
The moisture content of the powder blend is critical, as high moisture can lead to clumping, microbial growth, and poor compressibility during tableting. It is typically determined using a moisture analyzer or by a Loss on Drying (LOD) method.
Table 1: Summary of Pre-Compression Parameters for Tablet Powder
|
Angle of Repose |
Funnel method |
θ=tan−1(h/r) |
Flow properties |
<40° good flow |
|
Parameter |
Method |
Formula |
Significance |
Acceptable Limit |
|
Bulk Density (BD) |
Graduated cylinder |
BD=M/Vb |
Packing property |
Not applicable |
|
Tapped Density (TD) |
Tapping method |
TD=M/Vt |
Compressibility |
Not applicable |
|
Carr's Index |
Derived from BD & TD |
(TD−BD)/TD×100 |
Flow |
<20% good |
|
Hausner's Ratio |
Derived from BD & TD |
HR=TD/BD |
Flowability |
<1.25 good |
|
Moisture Content |
Moisture analyzer/LOD |
% weight loss after heating |
Prevents sticking & degradation |
2-5% (general limit) |
3.2 Post-Compression Parameters
Once tablets are compressed, a series of quality control tests are performed to ensure their uniformity, strength, and patient acceptability.
The appearance of a tablet is the first visual quality check. This includes observing its size, shape, color, odor, and surface finish to ensure it is free from defects like cracks, capping, or mottling.
This test ensures that each tablet contains the intended amount of drug by verifying a consistent weight across a batch. According to standard pharmacopeial methods, 20 tablets are weighed individually to calculate an average weight, and each tablet's weight is then compared against this average. The allowable percentage deviation depends on the average weight of the tablet, with stricter limits for heavier tablets2,18.

3.2.3 Hardness (Crushing Strength)
Tablet hardness is the force required to break a tablet by applying pressure diametrically. It's an important test because if a tablet is too soft, it may break during handling or packaging, while a tablet that's too hard may not disintegrate properly, leading to poor dissolution and bioavailability. Hardness is measured using instruments such as a Monsanto or Pfizer hardness tester19.

The thickness and diameter of a tablet are measured using a Vernier caliper or micrometer. Ensuring uniform size is important for patient acceptability and for proper packaging and dispensing. These dimensions are directly influenced by the die fill and the compression force used during manufacturing.
Friability measures a tablet's tendency to chip or break under mechanical stress, such as in a blister pack or bottle. The test is conducted using a Roche Friabilator, where 20 tablets are tumbled for 100 revolutions11. The percentage of weight loss is then calculated, with an accepted limit of less than 1% to pass the test.

This test determines the time it takes for a tablet to break up into small fragments in a specified liquid medium under controlled temperature conditions. The test is performed using a disintegration test apparatus. For immediate-release tablets, the disintegration time is typically less than 15 minutes, while for film-coated tablets it is less than 30 minutes. Enteric-coated tablets have a specific requirement to not disintegrate in gastric fluid but to disintegrate in intestinal fluid20.

The dissolution test is a critical in-vitro performance test that measures the rate and extent to which the drug dissolves from the tablet into a liquid medium. This test is a key indicator of a drug's bioavailability and is essential for regulatory approval. The most common apparatus used are the
USP Type I (Basket) and Type II (Paddle)11.

Content uniformity ensures that the active drug is evenly distributed throughout the batch and that each tablet contains the correct dose. This test involves analyzing 10 individual tablets using a validated assay method, such as HPLC or UV spectrophotometry. According to USP criteria, each unit must contain between 85-115% of the label claim, with a relative standard deviation (RSD) of 6% or less2,21.
Stability testing assesses a tablet's quality over time under specific environmental stress conditions, which helps determine its shelf life. Common conditions include accelerated stability studies ( 75% RH for 6 months) and long-term stability studies
(60% RH for 12-24 months)16,19. During these studies, parameters such as appearance, assay, dissolution, hardness, and friability are monitored to ensure no significant changes occur.

Table 2: Post-Compression Tests for Tablets
|
Parameter |
Instrument/Method |
Significance |
USP/IP Limit |
|
Appearance |
Visual inspection |
Acceptability, compliance |
Should be uniform |
|
Weight variation |
Weighing balance |
Ensures dose uniformity |
±5-10% |
|
Hardness |
Monsanto/Pfizer tester |
Tablet strength vs. disintegration |
4−10 kg/cm2 |
|
Thickness/Diameter |
Vernier caliper |
Uniformity in size |
±5% variation allowed |
|
Friability |
Roche friabilator |
Resistance to chipping |
≤1% |
|
Disintegration |
Disintegration apparatus |
Tablet breakdown time |
≤15−30 min (depends on type) |
|
Dissolution |
USP Basket/Paddle |
Drug release rate |
As per monograph |
|
Content uniformity |
Assay (UV/HPLC) |
Dose uniformity |
85-115% |
|
Stability |
Climatic chambers |
Shelf-life & storage |
No significant change |
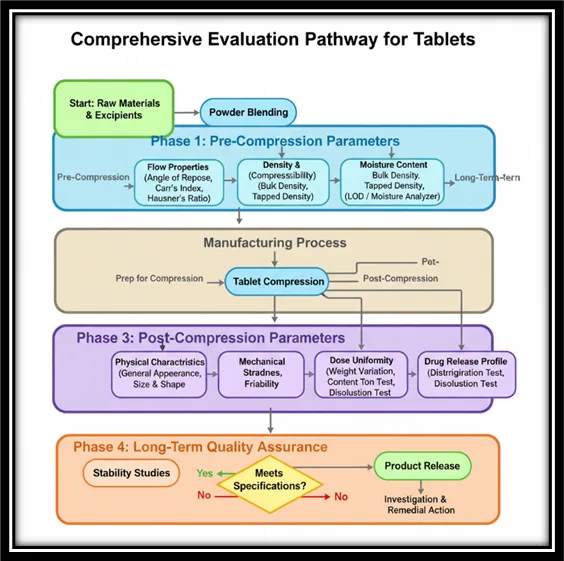
Figure 1 Comprehensive Evaluation Pathway for Tablet


4. Evaluation Tests for Capsules
Capsules are a widely used solid oral dosage form, available in hard and soft gelatin types. Thorough evaluation is crucial to ensure the safety, efficacy, and stability of the product before it is released to the market.
4.1 Pre-formulation and Pre-filling Evaluation
Before the capsules are filled, their components are carefully evaluated.
4.1.1 Capsule Shell Properties
The capsule shells themselves must be inspected for quality. A visual inspection is performed to ensure the shells are smooth, have a uniform color, and are free from cracks or dents. The moisture content of the shells is also a critical parameter; for hard gelatin shells, it should be maintained at 13-16% to ensure stability. This is typically measured using Karl Fischer titration. The dimensions and weight of the shells are also checked to ensure they are uniform, ranging from size 000 to 510,22.
4.1.2 Flow Properties of Fill Material
For hard gelatin capsules, the flow properties of the powder or granule fill material are essential to ensure accurate and uniform filling. The same pre-compression tests used for tablets—including angle of repose, bulk density, tapped density, Carr's index, and Hausner's ratio—are applied to the fill material4,6.
4.2 In-process and Post-filling Evaluation
After the capsules have been filled, a series of quality control tests are conducted on the finished product.
Finished capsules are visually inspected to confirm they are uniform, smooth, and free of any splits, dents, or sticking. This simple check is crucial for patient acceptability and for identifying any issues from the filling process.
The weight variation test is performed to ensure each capsule contains the intended amount of drug. The method involves weighing 20 capsules individually, emptying their contents, and then weighing the empty shells. The fill weight is then calculated and compared to the average2,18. The limits for weight variation in capsules are the same as for tablets.
This test ensures an even distribution of the drug, guaranteeing that each capsule contains the correct dose. Ten individual capsules are assayed using a method like UV or HPLC. According to USP criteria, each unit must fall within 85-115% of the label claim, with a relative standard deviation (RSD) of 6% or less21,22.
This test measures the time required for the capsule shell to break apart and release its contents. Using a USP disintegration tester, the capsules are submerged in a liquid medium, typically water at 37°C. For hard gelatin capsules, the limit is typically 30 minutes or less. Enteric-coated capsules have a specific requirement to not disintegrate in an acidic medium for two hours but to disintegrate in an intestinal medium20.
The dissolution test determines the rate and extent of drug release from the capsule. It is a critical performance test, performed using a USP Type I (basket) or Type II (paddle) apparatus in a specified buffer medium. The percentage of drug released is monitored over time11.
4.2.6 Moisture Permeation Test
This unique test evaluates how well the capsule shell resists environmental humidity. A capsule containing a color-changing desiccant is placed in a humidity chamber. Any change in the desiccant's color indicates that moisture has penetrated the shell10.
4.2.7 Mechanical Strength (for soft gelatin capsules)
For soft gelatin capsules, a compression test using a hardness tester is performed to ensure they can withstand the stresses of packaging and handling without breaking19,22.
Capsule shells, particularly those made of gelatin, can support microbial growth if not properly treated or stored. Therefore, microbial testing is conducted to check for total aerobic counts, molds, and pathogens, as specified by USP <61> and <62>2.
Stability studies are conducted on finished capsules under accelerated ( 75% RH) and long-term storage conditions to predict their shelf life. During these studies, key parameters are monitored, including microbial contamination, capsule brittleness, and drug potency and dissolution over time10,16.
Table 3: Capsule Evaluation Tests Summary
|
Parameter |
Instrument/Method |
Significance |
USP/IP Limit |
|
Appearance |
Visual inspection |
Acceptability, uniformity |
Should be uniform |
|
Shell moisture |
Karl Fischer titration |
Capsule stability |
13-16% |
|
Weight variation |
Weighing + emptying shell |
Dose uniformity |
±5-10% |
|
Content uniformity |
Assay (UV/HPLC) |
Dose accuracy |
85-115% |
|
Disintegration |
USP tester |
Capsule breakdown time |
≤30 min |
|
Dissolution |
USP basket/paddle |
Drug release profile |
As per monograph |
|
Moisture permeation |
Desiccant color test |
Packaging quality |
No visible change |
|
Mechanical strength |
Hardness tester |
Handling & transport resistance |
Should not break |
|
Microbial limit |
Plate count methods |
Safety |
As per USP <61>, <62> |
|
Stability |
Climatic chambers |
Shelf-life prediction |
No significant change |

Figure 3 Capsule
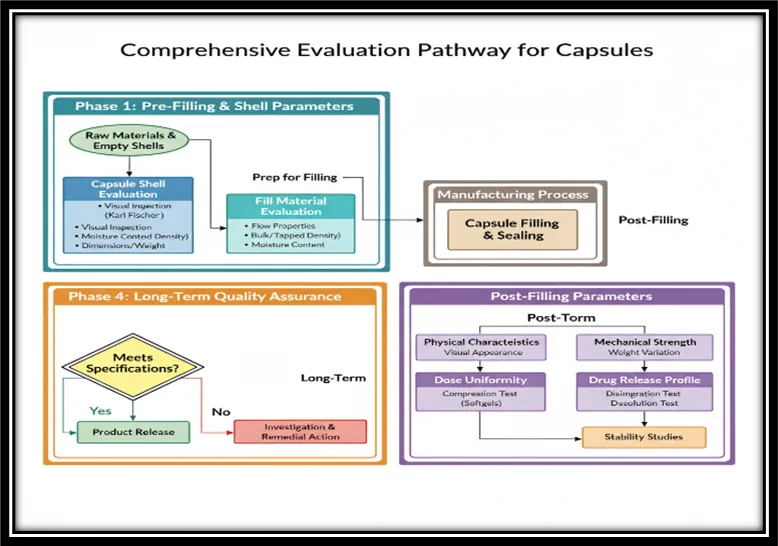
Figure 4 Comprehensive Evaluation Pathway for Capsul
5. Comparative Synthesis: Tablets vs Capsules
While both tablets and capsules serve the fundamental purpose of delivering oral medication, they differ significantly in their composition, manufacturing processes, and the specific quality control parameters required for their evaluation. A comparative synthesis highlights these differences and clarifies why distinct evaluation methods are necessary for each dosage form.
5.1 Formulation and Manufacturing Differences
Tablets are manufactured through a compression process where an active ingredient is combined with various excipients and compacted into a single, solid unit. This method requires the powder blend or granules to have specific flow properties and compressibility to ensure a consistent and high-quality final product. The formulation process can be challenging, as formulators must carefully balance the ingredients to achieve the desired physical properties, such as hardness, friability, and disintegration profile, while also ensuring the drug remains stable and bioavailable.
Capsules, on the other hand, are essentially pre-made shells that are filled with a drug substance. The filling process is generally simpler and offers greater flexibility in terms of the fill material, which can be a powder, granules, pellets, or even a liquid. Capsules are particularly useful for drugs that cannot withstand the high pressure of tablet compression, such as those that are heat-sensitive or have low melting points.
5.2 Evaluation and Quality Control
Many evaluation tests are shared between tablets and capsules, such as weight variation, content uniformity, and dissolution. However, the methods and significance can vary.
5.3 Biopharmaceutical and Patient Considerations
From a biopharmaceutical perspective, capsules are often associated with faster absorption and higher bioavailability compared to tablets because the capsule shell disintegrates quickly in the digestive tract, allowing the active ingredients to be released and absorbed more rapidly. Tablets, which must first disintegrate and then dissolve, can sometimes have a slower onset of action. However, advances in tablet formulation, such as the use of fast-disintegrating or orally disintegrating tablets (FDT/ODT), can help to overcome these limitations.
From the patient's perspective, capsules are often preferred because they are generally easier to swallow due to their smooth, uniform surface14. They also effectively mask the unpleasant tastes and odors of certain drugs. Tablets can be more difficult to swallow for some and may leave a bad aftertaste if not properly coated.
From a cost standpoint, tablets are generally less expensive to manufacture than capsules due to lower material costs and more efficient production processes. However, capsules offer a greater degree of formulation flexibility, which can be an advantage for certain drug properties or for niche markets.
Table 4: Comparative Evaluation of Tablets vs. Capsules
|
Parameter |
Tablets |
Capsules |
|
Physical Strength |
Tested for hardness and friability to prevent breaking or chipping during handling. |
Tested for mechanical strength and shell integrity to prevent deformation or leakage. |
|
Disintegration |
Solid matrix breaks apart into small fragments by superdisintegrants. |
Shell dissolves or ruptures to release contents; sensitive to shell material. |
|
Common Problems |
Capping and lamination due to poor compression. |
Brittleness and cross-linking due to moisture sensitivity. |
|
Moisture Permeation |
Not a primary concern for the solid form itself. |
Critical test to ensure the shell protects the fill material from environmental moisture. |
|
Manufacturing Cost |
Generally lower due to efficient compression. |
Generally higher due to material costs and filling process. |
|
Patient Acceptability |
Can be difficult to swallow; taste masking is often required. |
Generally easier to swallow; effectively masks unpleasant tastes and odors. |
6. Critical Appraisal & Practical Recommendations
The systematic evaluation of tablets and capsules, while guided by pharmacopeial standards, requires a critical appraisal of their methodologies and practical application in a real-world setting. This section highlights common challenges and provides best-practice recommendations for ensuring quality and regulatory compliance.
6.1 Common Pitfalls and Challenges
Despite standardized procedures, several common issues can arise during the evaluation and manufacturing of solid oral dosage forms.
Cross-linking and brittleness can occur in gelatin capsule shells due to improper storage conditions or interactions with certain ingredients, which can significantly affect the capsule's disintegration and dissolution profiles. Another issue is leakage in softgels, which can compromise the stability of the drug and affect the dose uniformity10,16,22.
6.2 Best-Practice Recommendations
To overcome these challenges and ensure robust product quality, the following recommendations are advised:
In essence, a critical appraisal of evaluation tests moves beyond simply checking boxes for regulatory compliance. It involves understanding the complex interplay between material properties, manufacturing processes, and the final product's performance. By adopting best practices and leveraging modern technologies, pharmaceutical companies can ensure dose accuracy, consistent release, and patient safety while also enabling efficient scale-up and technology transfer.
While the evaluation of tablets and capsules is a well-established science, several gaps and future directions in both technology and regulatory practice are shaping the future of pharmaceutical quality control. The field is rapidly moving from a traditional, batch-based testing model to a more proactive, patient-centric, and data-driven approach.
7.1 Gaps in Current Evaluation Practices
Despite the robustness of current pharmacopeial standards, some critical gaps and challenges remain:
7.2 Future Directions and Innovative Solutions
The pharmaceutical industry is embracing new technologies to address these gaps and create a more efficient and reliable evaluation process.
By adopting these future-focused strategies, the pharmaceutical industry can not only enhance the quality and safety of tablets and capsules but also streamline the manufacturing process, reduce costs, and accelerate the delivery of life-saving medicines to patients.
This review has thoroughly consolidated and appraised the essential evaluation tests for solid oral dosage forms, specifically tablets and capsules. From the initial pre-compression analysis of powder blends to the final stability studies of the finished product, it is clear that a rigorous and systematic approach to quality control is the cornerstone of modern pharmaceutical manufacturing. By ensuring that every product meets established pharmacopeial and regulatory standards, manufacturers can guarantee dose accuracy, consistent drug release, and patient safety.
The enduring dominance of tablets and capsules in the global market is a testament to their inherent advantages, including patient convenience, cost-effectiveness, and stability. However, their widespread use places a critical responsibility on the industry to maintain stringent quality. The review has highlighted that while both forms share some core evaluation parameters like weight variation and content uniformity, their unique characteristics necessitate distinct testing protocols. For tablets, key tests focus on mechanical strength, such as
hardness and friability, to ensure the compressed form withstands physical stress. Conversely, capsule evaluation places a greater emphasis on shell integrity and its susceptibility to moisture, which can lead to issues like brittleness or cross-linking.
A critical takeaway from this review is that a static, post-production quality control model is no longer sufficient. The pharmaceutical landscape is rapidly evolving, with a shift towards more proactive, in-process, and real-time evaluation methods. The integration of
Process Analytical Technology (PAT), including non-destructive techniques like Near-Infrared (NIR) spectroscopy and Raman spectroscopy, represents a significant leap forward. These technologies allow for continuous monitoring of critical quality attributes, enabling a more efficient and reliable manufacturing process. Furthermore, the advancement of
in-vitro/in-vivo correlation (IVIVC) is improving our ability to predict a drug's performance in the body, which can streamline development and reduce the need for extensive in-vivo studies.
In summary, the robust evaluation of tablets and capsules is a dynamic field that balances time-honored pharmacopeial requirements with innovative technologies. The move toward continuous manufacturing, advanced data analytics, and patient-centric formulations will define the future of quality control. By embracing these advancements, the industry can not only address common pitfalls like capping in tablets and cross-linking in capsules but also ensure that pharmaceutical products are consistently safe, effective, and of the highest quality for patients worldwide.





Manas Nikam, Monika Madibone, Rupali Pathre, Neha Pandit, Akash Navpute, Evaluation Tests for Tablets and Capsules: A Review of Solid Oral Dosage Forms, Int. J. of Pharm. Sci., 2026, Vol 4, Issue 5, 7612-7628, https://doi.org/10.5281/zenodo.20425899
 10.5281/zenodo.20425899
10.5281/zenodo.20425899