
We use cookies to ensure our website works properly and to personalise your experience. Cookies policy
SSS Indira college of pharmacy, Vishnupuri, Nanded, 431606, Maharashtra, India.
Orphan drugs are created on the specifications of rare diseases that attack very few individuals, although the diseases together represent a significant challenge to world health. Such therapies are still not developed easily, as they have a small group of patients, cost a lot to develop and the possibility of uncertainties on economic returns is high. Regulatory frameworks have thus played a pivotal role in leading to innovation and augment access to medicines against rare diseases.COVID-19 inevitably affected global regulatory systems, with stakey clinical trials, but with a surprising push towards a more adaptable method to approvals (emergency authorisations/ rolling reviews) and utilisation of real-world evidence. Notably, these adaptations provided hints to new and more effective methods of regulatory pathways - particularly required in orphan drug development.The current paper is a comparative post-COVID overview of the regulatory policies regarding orphan drugs in the United Kingdom and India. United Kingdom has developed a good tiered adaptive regulatory framework supported by Medicines and Healthcare products Regulatory Agency that is geared towards expedited access to new molecules. India is only beginning to achieve traction in terms of such policies as National Policy on Rare Diseases, but there is still no funding or infrastructure assistance, and the actual incentives are still paltry.All in all, the regulative adaptation in the two countries was similar in that there were major gaps in the policies implementation and access as effective treatments are being offered to the people. Expanding global coordination, solidifying regulatory frameworks and, most importantly in India, introducing focused incentives are key to increasing orphan drug development with opportunities translating into fairer access.
Orphan drugs are so named, that they are medicinal products that are indicated by the diagnosis, prevention or treatment of rare disease - that is, diseases that affect a few individuals compared against other members of a population but as a group pose significant burden on the health of the population. The development of orphan drugs was introduced into the world with a bang after the introduction of Orphan Drug Act in 1983, which offered a regulatory platform to encourage pharma companies to Develop treatments to treat diseases that scourged small groups of patients (Table 1). These historic regulations provided incentives such as market exclusivity, tax credits and fee waiving that propelled an unexplored field of drug discovery into a solid field of pharmaceutical research [1]. This prompted the establishment of these similar regulatory frameworks internationally, such as in the European Medicines Agency and other national agencies [2].
Rare diseases are not common per se, but they affect a big percentage of the global population at large. It is estimated that there are over 7000 rare diseases, which affect 300-400million people around the world [3]. A high proportion of most of these are chronic, progressive and life-threatening diseases that are genetically founded. This is particularly intense in India with huge population and a non-homogeneous collection of genes, where almost 70696 million individuals are estimated to have rare diseases [4]. Although the prevalence is high, it is challenging to diagnose and treat as there is little awareness, scarce healthcare facilities and lack of proven therapy [5].
Orphan drugs are scientific, economic and regulatory challenges in their nature. Although large-scale clinical trials cannot be conducted because of a small number of patients, the commercialization motives of pharmaceutical companies are further reduced by the high research and development expenses. Consequently, regulatory support is important to the development of orphan drugs. We need incentive mechanisms like market exclusivity, pathways for accelerated approvals, assistance with protocols and subsidies in order to drive innovation in this area [6]. There is an increasing attention by global regulatory bodies to adaptive, flexible frameworks to address the special needs presented by rare diseases.
Orphan drug regulatory frameworks had advanced internationally, but varied across the regions with the COVID-19 pandemic. Under the UK system, most of the regulatory framework was still in line with European Union frameworks via the European Medicines Agency and laid pathways to orphan designation and approval [2]. However, India itself was just in the process of establishing its regulatory infrastructure on such a concept and has a very low number of formal provisions on orphan-drug designation and incentives. The implementation of the New Drugs and Clinical Trials Rules (NDCTR) by Drugs Controller General of India in 2019 was a step in the right direction and even partially successful, however, implementation and gaps in policy were glaring [7].
The consequences of the COVID-19 pandemic, which first started in 2020, have had such a huge impact on both the healthcare systems and regulatory regimes in countries. The regulatory authorities injected an unheard-of leeway to hasten the creation and authorization of vaccines and therapeutics. There was increased expedited approval, reviewing on a roll, and use of real-world evidence became the norm [8]. These adaptive methods demonstrated that there was room to innovate regulation and signalled that it is in practice in various therapeutic domains, including orphan drugs.
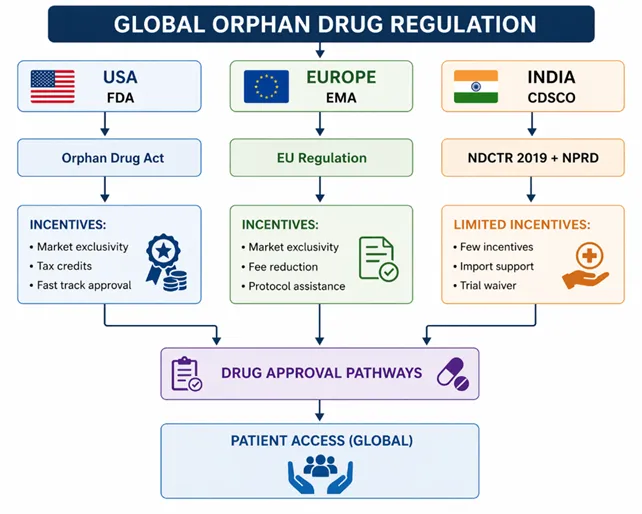
Figure 1: Global Orphan Drug Regulatory Framework and Approval Pathways
With the COVID pandemic, it is now more apparent that regulatory pathways of orphan drugs need to be reassessed and strengthened. The pandemic has highlighted the necessity of quick access to new therapies, well-developed health care systems on the national level, and international collaboration. Even for countries like the UK, Brexit and its regulatory independence have played a role in determining policy development, while steps to solidify rare disease policies in India exist via the National Policy for Rare Diseases 2021 (NPRD) [9].
Given the changing regulatory policies for orphan drugs in a post-COVID world, an extensive assessment is timely and needed. This review aims to critically scrutinize and juxtapose the recent regulatory changes in UK and India, challenges and opportunities, and to recommend future policy directions, which will enhance access of the orphan drugs to patients with the rare diseases.
1. Background on Orphan Drug Regulations (Pre COVID-19):
1.1 Global Regulatory Framework:
Prior to the onset of COVID-19, the regulation of orphan drugs had gravitated around frameworks implemented in some larger jurisdictions such as the United States and European Union. They exist by design to address the challenges of developing rare disease drugs, including small patient populations, costly research efforts and unpredictable returns on investment. It later on became a global benchmark towards spurring innovation and availing treatment of orphan diseases to the patients.
Orphan Drug Designation System of the US FDA:
Orphan Drug Act was the beginning of regulation of orphan drugs in the US and it is regarded as one of the most significant regulation frameworks ever written. This paradigm includes orphan designation by the U.S. Food and Drug Administration for drugs with an intended use in diseases affecting <200,000 people in the United States or that are unlikely to provide a profit [1].
The FDA orphan designation system is a framework that offers EPA regulatory assistance, scientific guidance and incentives that are available at an earlier phase of the drug development process. Sponsors can request an orphan designation at any stage of drug development prior to the submission of a marketing application. Orphan designation is especially important as it differs from marketing approval, but greatly eases the path toward potential development by lowering financial and regulatory hurdles [10].
One of the hallmark features of the FDA is its flexibility in regard to the requirement of the clinical trials. The limited groups of patients involved here refreeze their conception in alternative methodologies and endpoints, surrogate and adaptive. The large number of orphan drug approvals over the course of several decades in the US is, at least partly, due to this regulatory flexibility [11].
Framework of the European Union and Regulation by the EMA:
Orphan drugs in EU have a centralized regulations system under Regulation (EC) No. 141/ 2000 that is managed by the European Medicines Agency. Under EU law, a medicinal product can be termed as an orphan drug only when it is created to diagnose, prevent or treat a debilitating or life-threatening sickness to accomplish the same return on investment that the development of the product would otherwise have accomplished without its investment [12].
The Committee of Orphan Medicinal Products (COMP) of the EMA plays the central role in the assessment of orphan designation application. As is well known, just like the FDA system, sponsors also could make use of protocol assistance, as it provides scientific support on the study design and endpoints and regulatory provisions. The centralized procedure guarantees, that if approved, the drug can be marketed throughout all EU member states increasing the accessibility [13].
The EU system also enhances the idea of significant benefit to new orphan drugs, that is, there must be a clinically meaningful contribution to the existing treatments. This ensures that incentives are focused towards new, innovative treatment rather than other products with added marginal benefit [14].
Incentives for Orphan Drug Development:
It has one of its pillars in offering incentives to stimulate R&D in the regulation of orphan drugs around the world. Both the US and EU model offers numerous financial and regulation incentives tailored to cover the economic challenges of small markets associated with market access.
The greatest is market exclusivity. In the U.S., orphan drugs receive a seven-year exclusivity period upon approval meaning that no other products with the same indication can be obtained during the period. This exclusivity in the EU lasts not longer than ten years with possible extensions depending on various factors [1,12]. It gives a temporary monopoly and the sponsors will be able to break even on expenses.
The other important incentive is tax credits, particularly in the US where sponsors can claim tax credits on a significant amount of their clinical trial expenses. This will counterbalance the expenses of developing drugs in rare disease and offer financial incendivity to fund research in rare disease [15].
There are also cost savings of regulatory fees through waivers and abatements. Under the Prescription Drug User Fee Act (PDUFA), the FDA provides exemptions related to application fees and the EMA provides a reduction in scientific advice, inspections and marketing authorization applications, particularly to small and medium-sized enterprises (SMEs) [13].
Both regulatory systems are also characterized by grant funding programs, expedited approval programs and protocol assistance among others, and provide a supportive environment to orphan drug development that goes beyond standard market exclusivity regimes. These incentives have been effective in boosting the approvals of orphan drugs and in stimulating the development of therapeutics of rare diseases [6].
1.2 UK Regulatory Framework (Mixed EU Pre-Brexit):
Prior to the exit of the UK out of the EU, the UK had been a part of a harmonized regulatory framework, which was majorly dependent on the EU laws that regulated regulatory bodies in medicinal products, including orphan drugs. This coordination led to a consistent framework in drug approval criteria, access routes and incentive schemes among the member states, which allowed patients to gain access to new therapies to treat rare diseases at the right time.
Scope of the role of MHRA (Medicines and Healthcare products Regulatory Agency)
The Medicines and Healthcare products Regulatory Agency is the national competent regulator of medicines and medical devices in UK who regulates lifespan safety, quality and efficacy. Although orphan drug designation and centralized approvals were more of an EU concern, the MHRA played a vital role in pharmacovigilance, clinical trial approval and post-marketing approval[16].
MHRA was also an active participant in the regulations processes in the EU contributing the expertise of the EU agencies committees (European Medicines Agency (EMA)) that influenced the decision-making on orphan medicinal products. It also encouraged nationwide the adoption of EU specifications and policies that allowed approved orphan medications to be supplied to the UK health care delivery system [17].
Fitness to EMA Before Brexit:
Until Brexit, the UK was under the European Medicines Agency regulation. Gained orphan drug designation and marketing approval through the centralized process with EMA in Reg. (EC) No. 141/2000. Marketing approval might be approved by all the EU member states (including the UK) through the same regulatory procedure of approved orphan drugs without intermediate national approval [18].
This synergy had several advantages such as: One — faster approval processes, Two — expansion of geographical area of clinical trials and Three - homogeneity in regulatory standards. The UK too was able to capitalize on the EMA's incentive frameworks, such as ten years of market exclusivity and protocol facilitation with reductions in fees [13].
Simultaneously, however, the reliance on this centralized EU apparatus constrained the independent capacity of the UK to determine the policy in regard to orphan drugs. Large amounts of key decisions made on orphan designation; timing of approvals and regulatory requirements were majorly made at the EU level with national agencies playing a supportive and not directive role [19].
The Early Access to Medicines scheme (EAMS):
In 2014, UK announced its Early Access to Medicines Scheme (EAMS), a centralized scheme run by the MHRA, as an addition to the centralized EU framework. This initiative was the one that was to enable patients with life threatening illnesses or severely debilitating conditions to access investigational medicines before being approved in the market [20].
The EAMS evaluates medicines based on their benefit- risk and degree of unmet medical need. It is a two-step process with designation as a Promising Innovative Medicine (PIM) followed by a scientific opinion by the MHRA under the initiative. Such pathway is especially pertinent for orphan drugs, where access delay can drastically affect patient outcomes [21].
EAMS is among the earliest instances that provides flexibility and novel regulation to facilitate quicker access without debasing the safety and efficacy profiles. This created a model of the post-COVID regulatory changes that demonstrated the possibilities of expedited paths to address the most urgent medical needs [20].
1.3 Indian Regulatory Framework:
One major difference between the regulatory approaches in the US and EU, compared to India (prior to COVID-19), was comparatively limited framework for orphan drugs. Although a lot had been done to strengthen the 'general' pharmaceutical regulatory system, there were very few specific provisions regarding orphan drugs and rare diseases.
Central Drugs Standard Control Organization (CDSCO):
The national regulatory authority of drugs approval, clinical trials and pharmaceutical standards in India is the Central Drugs Standard Control Organization (CDSCO). CDSCO operates as an arm of Directors General Health Services that works in collaboration with the centre and the state government to guarantee safety, efficacy and quality of drugs currently being marketed in the country [22].
CM:PD: The regulatory framework of CDSCO has never had a specific route to orphan drug designation before COVID-19. Until the New Drugs and Clinical Trials Rules (NDCTR) came into force in 2019. These regulations also offered certain flexibilities such as a faster approval routes and exemptions of local requirements of clinical trials in conditions which would be relevant to the orphan drugs [7].
Introduction- National Policy for rare diseases (NPRD 2021):
The National Policy for Rare Diseases, although not formalized until 2021, is a reflection of the policy that had been developed prior to and during COVID-19. The NPRD is designed to help overcome major hurdles related to rare diseases such as diagnosis, ability to treat the patients and the overall cost of that care.
It centres on the establishment of Centres of Excellence, national registry and subsidy in the treatment of the select rare diseases. What is more important, it accentuates the urgency to develop research and domestic production of orphan drugs [9].
Nevertheless, in these regards, there have been some significant accomplishments, but the NPRD does not offer a complete regulatory framework, similar to the US or EU frameworks. It is now more concerned with healthcare provision and patient care than with formal incentives in the pharmaceutical industries.
A byzantine Orphan Drug pre-COVID:
Prior to the pandemic, India lacked a separate regulatory route to orphan drug designation, approval and post-marketing support. Unlike the US and EU, no special incentives like exclusivity in the market, tax reduction or dedicated grant program was dedicated to the development of orphan drugs were provided [23].
Therefore, due to this uncoordinated policy, this industry has had to deal with a number of pitfalls, including minimal industry participation, dependence on importation of therapies and high cost of treatment to the patient. This was compounded by a lack of an official rare disease register and the lack of diagnostic capacity, which ultimately rendered a unifying policy response virtually impossible [24].
This rendered orphan drugs accessible to just a handful of patients in India and it was almost economically unattainable to a group of patients as financial and logistical constraints towered over such treatment. These restrictions intensified the need to reform regulations, which became increasingly popular after the COVID as health policymakers saw the possible merits of resilient and adaptive health systems.
2. The COVID-19 Impacts on Regulatory Systems:
2.1 General Impact:
In late 2019, the COVID-19 pandemic was identified and spread around the world and unprecedented waves of disruption through disruptions to healthcare systems (HS) & regulatory processes (RP) worldwide. Regulatory agencies were forced to quickly revise frameworks to keep drug development on track and meet the pressing public health issues. These developments had regulatory implications, such as with respect to orphan drug development.
Table 1: Impact of COVID-19 on Drug Regulatory Systems
|
Aspect |
Pre-COVID |
Post-COVID |
|
Clinical Trials |
On-site, rigid |
Remote, decentralized |
|
Approval Process |
Time-consuming |
Accelerated (rolling review) |
|
Data Usage |
Traditional clinical data |
Real-world evidence (RWE) |
|
Monitoring |
Physical visits |
Digital/remote monitoring |
|
Regulatory Approach |
Conservative |
Flexible & adaptive |
Interruption of Clinical Trials:
One of the most direct effects the pandemic had on the active clinical trials was the spread of the pandemic. Travel and reallocation of health care to focus on COVID-19, lockdowns, prevented recruitment of patients in need of emergent treatments, interrupted protocols and data collection [25]. Protocol deviations and missing data occurred as many of the sites used to conduct clinical trials were abandoned and subjects could not easily access the planned visits [26].
Regulatory agencies (e.g. the U.S. Food and Drug Administration and the European Medicines Agency) published guidance documents on such delays in product manufacturing to assist in reducing these problems [13A]. These recommendations allowed the amendments of protocols, off-site monitoring and other ways of data collection to maintain the integrity of the trial but focus on the safety of the patients [27]. These upheavals revealed the vulnerability of traditional clinical trial models, and demonstrated the need to have more adaptive and resilient ones.
This allows the process of regulatory pathway acceleration:
Since the world was in urgent need of COVID-19 treatment drugs and vaccines, regulations bodies took expedited approval procedures. Two approaches that were employed towards this were rolling reviews and conditional approvals that resulted in cutting down of processes that previously took years and parallel assessment [28].
This resulted in unparalleled cooperation and effectiveness of regulatory authorities, which enabled conducting more quick reviews without compromising the bar of safety and efficacy. Such expedited routes offered prompt access to the COVID-19 armamentarium and they also created a paradigm, which can be potentially applied to other medical fields, including those of rare diseases [29].
Digitalization, Remote Monitoring:
Covid-19 was the stimulus to accelerate the development of digital technologies in regulation and clinical studies. Devices like telemedicine, remote monitoring and decentralized clinical trials were required to maintain the continuity of studies [30]. Electronic informed consent (e-consent) and virtual site visits were also said to be used and the use of wearable health technologies in order to record real-time data on patients [31].
The regulatory agencies supported these innovations by informing us in general how to exploit digital tools and remain in line with the data integrity and privacy requirements. The shift to digitalization not only offered prompt solutions to the existing challenges and limitations but also demonstrated a higher level of efficiency in trials, reduced the burden of involvement on patients and better representation of different populations in the long run [30].
2.2 Lessons Learned:
These lessons were also important to the future of policy, particularly on the topic of orphan drug regulation as the COVID-19pandemic had taught us the possibilities and issues with our current regulatory frameworks.
Emergency Approval (e.g., Vaccines):
The COVID-19 pandemic saw several interesting regulatory reactions, including the use of emergency authorization tools of vaccines and therapeutics. To illustrate, Pfizer BioNTech and Moderna issued vaccines that were approved under emergency use (EUA) based on interim clinical data that demonstrated safety and efficacy [32].
These approvals required an adaptive trial design, live analysis and close interaction between the regulatory agencies and the developers. These effective methods demonstrated the fact that it is possible to implement quick ways without the compromise of needed scientific rigor with robust post-marketing surveillance systems [33].
Findings: There is flexibility in Regulatory Decision-Making:
You would see it during the pandemic; they need flexibility on regulations in addressing public health emergencies. Risk-based approaches were embraced by authorities, enabling conditional approvals, the use of surrogate endpoints and acceptance of real-world evidence (RWE) [34].
Such flexibility enabled regulators to ease the timely access and yet had to consider the safety and efficacy. It also promoted the element of innovation in terms of designing clinical trials and data analysis. These adaptive methods are especially applicable to, and should be considered in the context of orphan drugs, where the generation of evidence is often constrained because of small patient populations [35].
The Emergency to Rapid Therapy:
COVID-19 underscored the necessity to enable the access of high interest therapies when severity occurs. You really need to put to the fore that waiting until medication is given the green light can be life-death consequences when it comes to people with life altering ailments. The regulatory regimes revealed that expedited process, global cooperation and proactive policy frameworks would go a long way in reducing approval times [36].
This lesson is particularly applicable to orphan drugs, in which patients still have to endure prolonged treatment access. A regulatory framework has been the place where patient access has been high during the pandemic experience as they attempt to not compromise standards of safety and efficacy [37].
3. Since COVID Regulatory Development in UK:
In 2018, the United Kingdom has entered a new phase of the regulation of medicinal products in general, and orphan drugs, in particular. After quitting the EU and the experience during the pandemic, Britain has introduced a more permissive, innovation-focused regulatory environment with the ambition of longer-term access to patients in standards of safety and efficacy (and getting experience in the field).
3.1 Reforms and Policy changes:
MHRA Independence Post-Brexit:
Brexit was finally concluded in 2021 and the regulation of medicines and medical devices in the UK was fully transferred to the Medicines and Healthcare products Regulatory Agency. This was a change in England (EU procedures) into a national procedure and rules [38].
Following Brexit, the MHRA could develop and implement its approval pathways that can be more adaptable with regards to fulfilling new healthcare requirements. It put in place new regulatory orchestration procedures involving reliance pathways (e.g., acceptance of approvals by known trusted foreign regulators), and rolling reviews [39].
This independence provided for a much more flexible regulatory model, especially when it comes to rapid approvals of vaccines and therapeutics as seen during the COVID pandemic.
Introduction of the Innovative licensing and Access Pathway (ILAP):
In 2021, the UK also launched the Innovative Licensing and Access Pathway (ILAP) pathway in order to enable early access to new treatments. The ILAP should hasten the process of developing and approving novel therapies, even rare diseases [40].
The process brings the activity of a variety of organisations between the MHRA and the National Institute of Health and Care Excellence (NICE), down to that of NHS, into alignment so that this is a coordinated pathway to patient access, starting with early development. One of the key aspects of ILAP is the so-called Innovation Passport, which enables innovative products to have a more favourable access to regulatory, as well as scientific assistance [41].
By having a continuous dialogue with the developers and regulators, ILAP can streamline clinical trial design and determine upfront difficulties and set a common ground in terms of evidence requirements. Such a model of cooperation assists in enabling lifecycle-based regulation, and collaborates exceptionally well with orphan drugs that are in most cases fraught with enormous development and development challenges.
Faster Approval Pathways:
Based on the regulatory approaches established during the pandemic, it hastened its approvals process to reduce time-to-market of innovative therapies. This comprises rolling reviews, conditional approvals and sped up evaluation pathways [42].
By evaluating data and devices when the first pieces of evidence are available to regulators - as opposed to waiting for sponsors to file full basic submissions - these mechanisms can dramatically decrease approval timelines. Such methods, which are preferred, are fundamental in maintaining the rate of drug development business and enabling patients with severe unmet medical need to gain access on time [43].
These expedited routes are specifically relevant to orphan drugs whose approval process can save lives and deaths of patients with rare diseases who may have a putatively useful medicine.
3.2 Incentives for Orphan Drugs:
Market Exclusivity:
In the UK, market exclusivity is a suitable motivation in the development of orphan drugs. Following Brexit, the UK maintained a similar system to European, which offers UP up to 10 years of exclusive rights to orphan drugs in the market [44].
It implies that no other comparable product is allowed to be authorized with the same suggestion within the protected timeframe so that sponsors could recover research and-development. This incentive has been renewed which goes to show a commitment by the UK to further promote a competitive environment that fosters orphan drug innovation.
Fee Reductions:
In case you are a qualified and can take advantage of regulatory services by the MHRA, there are fee reductions and waivers on certain services based on the type of service and more so on SMEs and developers of innovative therapies. These cuts are associated with scientific recommendations, inspections and applications of marketing authorizations [45].
Such and other financial incentives are significant in alleviating the economic barriers to developing orphan drugs which is usually costly with unguaranteed returns due to the small number of patients.
Support for SMEs and Innovation:
The UK regulatory landscape is highly geared toward promoting innovation and SMEs. Examples include innovation offices, regulatory guidance programmes and funding support tools to spur early development of such therapies [46].
Programs like ILAP among others are tailored to help steer developers through scientific guidance, regulatory direction and approval process. Through orphan drugs, innovation is also a vital factor since most of the first-in-class innovations are brought to the market by smaller biotechnology companies.
3.3 Challenges:
Divergence from EMA:
Regulatory divergence between the UK and EU, which is likely to be one of the greatest problems in the post-Brexit set-up. Flexibility is provided by independence, but it is also subject to various approval requirements, various timelines and various standards than the European Medicines Agency [47].
This inconsistency will make the backgrounds of pharmaceutical companies trying to bring products to the two markets more challenging and may lead to the duplication of the efforts and increase in expenses. It would also jeopardise the UK as a major launching state of new treatments.
Limited Patient Population:
The UK has a low population when compared to the EU and this hinders the development and commercialisation of orphan drugs. Few patients can impact on recruitment, the generation of data and ultimately market viability for clinical trials [48].
This limitation can reduce the motivation to encourage pharmaceutical firms to target the UK particularly when larger markets are indicating greater commercial returns. To meet this challenge, new strategies − including global cooperation and real-world evidence − need to be employed.
Problems with market access (NHS If involved in price setting/reimbursement process):
Pricing and reimbursement continue to be key barriers to access for orphan drugs in the UK. Assessing the cost-effectiveness of new therapies is an important role of the National Health Service (NHS) and NICE [49].
The price of orphan drugs is usually very high due to the fact that there are only a limited number of patients and the processes used to manufacture them are complicated. This can complicate the decision of reimbursement which could delay patient access despite regulatory approval (50).
The cost-versus-innovation balance is still a policy issue and regulatory bodies, health care providers and industry stakeholders should collaborate.
4. Regulatory Evolution in India, Post-COVID:
THE COVID-19 PANDEMIC A Catalyst to regulatory reforms in INDIA The COVID-19 pandemic has presented a catalyst to the regulatory reforms that are long overdue to have a healthcare system that is responsive, flexible and patient cantered. India has gone a long way to establish a regulatory framework in regard to rare disease and orphan drug after the pandemic. As the policy and regulatory support is being developed, the development of an entire system competing with the incumbents internationally remains a challenge to be realised.
4.1 Policy Developments:
Strengthening of NPRD 2021:
THE NATIONAL POLICY ON Rare Diseases is a milestone in the management of the rare diseases in India. It was implemented long before the pandemic began, yet its application and emphasis on operations have become quite strong during the post-COVID period [9].
Orthotopic Diaphragm Transplantation: The policy puts access to a diagnosis and treatment in the light by developing Centres of Excellence (CoE), a national registry of rare diseases, and providing funding support in the case of some conditions. It does so by classifying rare diseases into certain groups based on the presence of a treatment and the cost implication; enabling policy intervention [51].
Another aspect of NPRD 2021 is its focus on indigenous research and the local manufacturing of orphan drugs which is in line with India’s more Braun “make In Indian” initiative. Although these aspects are not negligible the policy remains predominantly delivery-oriented and patient-oriented with insufficient incentive to create new pharmaceuticals.
The New Drugs and Clinical Trials Rules (NDCTR 2019) were updated:
One such move that gained a lot of importance especially during and after the COVID -19 pandemic was the operationalisation of the New Drugs and Clinical Trials Rules, 2019 (NDCTR) by Central Drugs Standard Control Organization. Such regulations included the faster drug approval and better regulatory oversight [7].
Following COVID NDCTR provisions have been increased to accelerate approvals (of drugs to meet unmet medical needs) more frequently. It offers expedited pathways of approval, reduced the time of reviews, and flexibility in the requirements of clinical trials under specific circumstances [52].
Although NDCTR is a foundation of regulatory flexibility, lack of an orphan drug system, unlike that in regimes in developed nations, makes NDCTR different.
Formalization of the definition of Orphan Drug:
Defining orphan drugs has been a big move in India. An orphan drug is a medication destined to treat a disease that has an incidence not excessively above 500,000 individuals in India as stipulated under NDCTR 2019 [7].
Such definition applies to making regulatory decisions and policy. It takes India into the same line with the world practices but the degree is lower than that applied in the US and the European Union. The formalisation required to establish effective incentives and regulatory avenues to orphan drugs in the future is the setting up of a clear definition [23].
4.2 Regulatory Support:
Fast-Track Approvals:
To mitigate severe shortages of essential medicines during the pandemic, Indian regulators used fast-track approval pathways. Such strategies have continued in the aftermath of COVID in particular of the drugs used to treat serious or life-threatening diseases such as rare diseases [53].
Faster approval processes provide a shorter review time on applications that are frequently based on less data concerning limited but compelling clinical evidence. This can be particularly handy when there is a small number of patients and the clinical trial is potentially too big to conduct.
Local pgx trials waiver (in some Cases):
Local clinical trials can be waived under specific circumstances under the NDCTR framework, such as when the drug is approved and marketed in other countries where there is a sound regulatory system [52]source].
The provision saves a lot of time and money involved in the approval of drugs in India and enables faster availability of international approved therapies. This can be flexible and is particularly helpful in the development of orphan drugs where local trials can be challenging because there might be few patients [54].
Import Facilitation:
In response to the scarcity of orphan drugs available domestically in India, steps have been taken to ease importation of necessary therapies. Provisions of law facilitate the legalization of unapproved medicines for personal use or with access through special permissions from authorities [55].
The administration also acted to hasten the process of importing and eliminate red tape on some life-saving drugs. They provide an important source of domestically-manufactured yet to be manufactured rare disease treatments, in which there are few treatment options.
5. Comparative Analysis: UK vs India
5.1 Regulatory Framework Comparison:
Structured vs Emerging System:
An ancient coordination with the European Union and post-Brexit reforms that institutionalized a framework turned it into well-organized and mature orphan drugs regulatory system in the United Kingdom (UK). The Medicines and Healthcare products Regulatory Agency works within a well-defined legal/procedural framework, that incorporates orphan designation, provisions of market exclusivity, coordinated access pathways [57].
Indians’ regulation system is, on the other hand, still in its infancy. National Policy for Rare Diseases, and New Drugs and Clinical Trials Rules (NDCTR) 2019 have rolled out some of the key reforms however there is an empty space on a focused functional orphan drug framework [9].
This disparity reflects regional disparities in healthcare facilities, priority and economical ability. Where, UK framework urges innovation and access velocity whereas India is still occupied with building the fundamental capabilities.
Regulatory Agency Role:
In the UK, medicines are regulated under the auspices of an independent government body, the Medicines and Healthcare products Regulatory Agency (MHRA) that has a central and unique role in working with other institutions including but not limited to: National Institute for Health and Care Excellence (NICE); the National Health Service. It is a combined strategy that aligns regulatory acceptance, health technology assessment and patient access [58].
By contrast in India, the approvals of the drugs and the execution of the clinical trials are generally supported by the Central Drugs Standard Control Organization (CDSCO) resulting in variations 【6 source】. Conversely, integration between regulatory, reimbursement and health care delivery systems is less. The multistakeholder aspect of policy implementation has been one of the causes of fragmentation and delay [59].
Table 2: Comparative Analysis of Orphan Drug Regulatory Systems in the UK and India
|
Parameter |
United Kingdom |
India |
|
Regulatory Authority |
MHRA |
CDSCO |
|
System Type |
Structured & mature |
Emerging |
|
Approval Speed |
Fast (ILAP, rolling review) |
Moderate |
|
Clinical Trial Flexibility |
High |
Limited |
|
Incentives |
Strong (exclusivity, fee reduction) |
Weak |
|
Funding Support |
Available |
Limited |
|
Patient Access |
NHS supported |
Out-of-pocket burden |
|
Policy Framework |
Well defined |
Developing |
5.2 Approval Pathways:
Speed and Flexibility:
Specifically, UK has incorporated very accommodative and speedy approval protocols in what has now been the post-COVID era. The mechanisms, like rolling reviews, conditional approvals, and the Innovative licensing and access pathway (ILAP) [40], could help to evaluate and approve innovative therapies including orphan drugs in a timelier manner.
India has also taken measures to expedite approvals particularly under the NDCTR provisions. However, a relatively low flexibility and efficiency is still caused by the regulatory capacity and complexity of the procedures [7].
Clinical Trial Requirements:
There exists a huge difference in the requirements in both countries of clinical trials concerning orphan drugs. The approach of the UK is patient-centric, scientifically adaptable to enable adaptive trial design, surrogate endpoints, and even be dependent on international data [43].
In some cases, where adequate data of other jurisdictions is accessible, India has established procedures to forego local clinical trials. However, these provisions are applied selectively and might not be applied uniformly[7].
Contrary to the UK system, lack of adequate infrastructure and patient registries in India affects the ability to conduct robust clinical trials in India on rare diseases.
5.3 Incentives and Support:
Financial Incentives (UK Stronger):
UK offers a wide range of financial and regulatory incentives to promote the development of orphan drugs. Which fall into, e.g. market exclusivity, fee-reductions, scientific advice and specific support of small and medium sized enterprises (SMEs) [46].
Such incentives enable innovation and investment that is supportive to development of drugs against rare diseases by pharmaceutical industry. Institutional funding mechanisms and public-private partnerships support this ecosystem.
India Policy Gaps:
In comparison, there is no complete incentive system in place in India to develop orphan drugs. The policies, such as NPRD 2021, assist patients, yet provide the pharmaceutical companies with almost no incentive directly [9].
Such absence of policy tools such as tax credit, Germaine grants and long market exclusivity renders the Indian market unattractive to develop an orphan drug. The consequence of such a loophole is that there is a significant importation of therapies in foreign countries and less developed ones locally [23].
5.4 Accessibility and Affordability:
Out of Pocket Burden vs. NHS reimbursement in India:
The UK and India are least similar in terms of accessibility and affordability. The UK has a universal health care system via the National Health Service and reimbursement decisions are based on cost-effectiveness evaluations done by NICE [58].
Orphan drugs are all too expensive, yet regular reimbursement frameworks and special financing initiatives make them accessible to patients. The clinical benefit and cost are also balanced using managed access agreements and risk-sharing schemes [50].
Health spending in India is mainly out-of-pocket, placing a lot of financial strain on patients and their families [60]. The prohibitive price of orphan drugs and lack of insurance coverage and funding source restrict the treatment to the luckiest.
Despite NPRD 2021 considering financial impact to certain diseases, access is limited or constrained to the extent that most patients are left with no option but to keep facing disproportionate barriers to life-saving therapies.
6. Economic and Ethical Issues:
The controversial issues in the development and regulation for orphan drugs have garnered more attention given their ethical and economic nature in the COVID post-era. Despite the regulatory incentives in place to encourage innovation, there exist challenges on affordability, sustainable access and ethical clinical research. These apprehensions are especially pertinent in the area of rare disease; wherein patient populations are few and treatment options also limited.
6.1 Orphan Drugs are expensive:
Orphan drugs are priced high since number of patients is small, development is expensive and it must recover these expenses at market extremities. Some orphan drugs can cost a significantly higher price to patients than traditional treatment, with some even costing hundreds of thousands of dollars annually [61].
Retracted to promote innovation, there is a need to have market exclusivity, fee waivers, expedited approvals but they can potentially affect high pricing during the market exclusivity period as there is less competition [1]. Moreover, complexity in the manufacturing processes, especially in biologics and gene therapy products, adds to the cost [6].
High-income countries (including the UK; pricing difficulty is tempered because of centralized health systems; such as National Health Services, which negotiate prices, and cost-effectiveness reviews) are to some extent able to overcome pricing difficulties. However, the financing provided to the high-cost therapies should be sustainable, despite the presence of such systems [70].
In comparison, the high cost of orphan drugs is an extreme limit to the accessibility of patients in India and other countries with out-of-pocket healthcare spending. Due to such economic expenses, most patients tend to postpone treatment or in some cases, they end up not receiving treatment [60].
6.2 Equity in Access:
Availability of orphan medicine is popularly viewed as an urgent ethical concern because to a large extent it is a disparity between high- and low- to middle-income countries (LMICs). Patients in developed countries can count on well-developed healthcare systems and reimbursement systems, but those in developing countries face tremendous burdens in accessing life-saving therapies [62].
The report additionally mentions stark inequalities in health within countries which are related to socioeconomic status, geography and access to healthcare infrastructure. In the United Kingdom, equitable access is supported by publicly-funded healthcare and systematic reimbursement trackways. Access can still be subject to cost-effectiveness and budget constraints [70].
In India, there is low insurance coverage which creates more inequities against noncommunicable diseases because of low health infrastructure and lack of diagnostic facilities. Though there are policies to encourage access like the National Policy on Rare Diseases that facilitates access through cost and patient assistance programs, to enable the formation of rare disease centres [9], these efforts have not propagated.
This creates a need to balance carefully both methods of encouraging innovation and access-promoting policies, particularly of underprivileged groups.
6.3 Ethical Consequences of Population Trials of Limited Population:
The special ethics in the case of orphan drugs pertains to the clinical trials that have been conducted to test such medications, since trials are done on a very small patient group. Because of the challenges of the implementation of the traditional randomized controlled trials (RCTs) these treatments are commonly evaluated through other study designs like single-arm trials, adaptive trials and surrogate endpoint [63].
Such methodological adjustment makes one question the quality and useability of clinical evidence. The uncertainty on safety and efficacy could be magnified by small sample sizes and patients are exposed to the risks of therapies that have not been sufficiently assessed as being safe and effective [64].
In addition, the trial design does have ethical issues like the placebo control in the absence of standard treatment. Refusing a potential treatment that could help patients, under such circumstances, can thus perhaps be argued to be unethical. To overcome these hurdles, regulators have been less strict on the front of trial design and necessity of well-designed post-marketing surveillance and collection of real-world evidence [34].
Another possible area of concern was that too many patients would be exploited by being offered a treatment option that they would otherwise not receive and that they would be more willing to be used in trials. There is therefore a need to ensure that the research conducted on orphan drugs is conducted with the highest ethical standards, with an informed consent and transparency policy and safeguarding the patients [65].
7. Future Directions:
The orphan agenda of altering regulation in the post-COVID era will progressively demand an out-of-the-box mindset that will ideally address the gaps left in accessibility due to innovation and enhance equity in the world. Although the regulatory maturity in the United Kingdom and India is different, still, there are prospective ways of further convergence in a collaborative, data-driven and patient-centric manner. The following areas of priority define some significant policy and regulatory directions in the future.
Global Requirement of Unity:
The significant barrier to the effective development and access to orphan drugs is that not all jurisdictions have harmonized regulatory standards. Although TCM is capable of playing a critical and transformative role in complementing modern Western forms of medicine, the absence of uniformity in the definition of the products of TCM, approval regulations, clinical trials requirements and incentive system, contribute to overlapping efforts and even more delay to market [66].
Or, an international convergence of standards and mutual recognition agreements between regulators can be achieved with the help of bodies like the World Health Organization, European Medicines Agency and national agencies. First, consistency of the requirements of the orphan drug designation and data would decrease the regulatory load and accelerate approvals in multiple regions [67].
The model of global cooperation in the development of vaccines in the (not only but also) post-COVID period could be a platform on which the attainment of the joint regulation could be expanded into the area of rare diseases development and orphan drugs development.
Public-Private Partnerships:
One of the keys to bypass the economics and scientific hurdles to produce orphan drugs is public and private partnership (PPP). Given that there are few commercial incentives that can encourage collaborative effort between government, academic and pharmaceutical companies, risk reduction can be achieved by way of resources and expertise scarcity [68].
E.g., synergies, such as a joint funding program, research networks, joint innovation hubs, can assist in enhancing the discovery and development of drugs in rare diseases. Such partnerships frameworks are already available in the UK that bring together regulatory bodies and health care systems with industry partners.
Bringing strong public and private partnerships (PPP) may form an important element in promoting indigenous research, reducing dependence on imports and making available cheap treatments to India. Funding, infrastructure, and policy incentives from the government are necessary for these collaborations to continue.
Building on Rare Disease Registries:
The establishment of stronger rare disease registries, which are part and parcel of diagnosis, research and regulatory decision-making will benefit everyone. They are useful databases of resourceful epidemiological information, facilitate clinical trials recruitment and generation of real-world evidence (RWE) [69].
Registries facilitated epidemiological data of disease burden, natural history and response to treatment in developed systems. Enhancing the connection between registries and UK health care systems has contributed to policymaking using the data.
It is remarkable that India has already started to develop a national rare disease registry according to the National Policy on Rare Diseases, but the issue of data standardization, coverage and integration with clinical infrastructure is a significant challenge [9]. The above-mentioned registries would require enhancement so that they can identify patients, conduct research and enable evidence-based control.
India Recommendation Policy:
Therefore, to establish a sustainable orphan drugs ecosystem, it is important that the Indian government should consider an absolute policy change that addresses the existing shortcomings when it comes to regulation, economic incentives and care delivery. Key recommendations include:
The measures will also guarantee that the access to the treatment is better, and India becomes a new participant in the development of orphan drugs [23].
Innovation of Orphan Drugs in the UK and its role in Global Innovation:
The UK is well positioned to lead the pack in innovation in orphan drugs internationally with a well-established regulatory system, intertwined research ecosystem and integrated healthcare system. To exemplify this desire to help people have access to innovative treatments, the Medicines and Healthcare products Regulatory Agency has been making several efforts such as: Innovative licensing and access pathway (ILAP) [40].
UK can solidify its dominance in the world and the future success of its life sciences industry by forming global partnerships, conducting multi-country clinical trials and shaping harmonised regulatory frameworks. Moreover, its emphasis on the real-life evidence, flexible trial designs, and patient-centred methods may serve as an example to the rest of the world.
As the UK regulatory agility and science ability remains, the UK has a genuine chance to remain an engine to orphan drug innovation, as well as to assist others with access to rare disease.
CONCLUSION
Results This review is a summary of a few of the changes that are in the process of being implemented (or already implemented) to the regulatory environment (especially regarding orphan drugs) in this post-COVID world through the comparative paradigm of the UK vs. India. In our analysis of the implementation of the regulatory frameworks in the Netherlands and Finland, we find that although both countries have made significant progress in strengthening their regulatory frameworks, there are still glaring gaps in terms of structure, efficiency and accessibility. The United Kingdom and respectively Medicines and Healthcare products Regulatory Agency (MHRA), have come up with a clear and innovation-focused framework, characterized by expedited approval procedures, strong incentive initiatives and combined health care supports. In India, however, where big steps have been taken through the introduction of the National Policy on Rare Diseases, there are still steps to be made with limited funding, inadequacy of infrastructure and incentives to encourage orphan drug development within the system.
This COVID-19 pandemic has shown over the last few years that flexible and adaptable regulatory systems are essential. The instruments of speeding up approval, digitalization and greater collaboration that shown by regulators in the pandemic may be used to enhance the orphan drug regulation. They can reduce the developmental cycles and enhance accessibility of patients especially in rare diseases that are presenting life-threatening symptoms.
But while orphan drug policies evolve, they need to strike the right balance between fostering innovation and guaranteeing access. The incentives are essential to motivate research and development, yet they must not lead to high prices and restrictions on the access to specific territories. This implies that policymakers must balance it around both affordability and equitable access and sustainable health systems.
Flexibility in regulations, global co-ordination and narrowly focused but comprehensive policy reforms in developing systems like those in India will be necessary to promote orphan drug development and give patients with rare diseases a timely and fair access to the most effective treatments.
REFERENCES




Rohit Muneshwa, Dr. Vijay Navghare, Dr. Suryakant Jadhav, Uday Gaikwad, Evolving Regulatory Policies for Orphan Drugs Post-COVID-19: UK and Indian Perspective, Int. J. of Pharm. Sci., 2026, Vol 4, Issue 5, 7248-7267, https://doi.org/10.5281/zenodo.20407102
 10.5281/zenodo.20407102
10.5281/zenodo.20407102