
We use cookies to ensure our website works properly and to personalise your experience. Cookies policy
Department of Pharmaceutical Quality Assurance, Rashtrasant Janardhan Swami College of Pharmacy, Kokamthan, Kopargaon, Ahmednagar, 423601, Maharashtra, India
Quality by Design (QbD) has emerged as a systematic, science-based approach for improving pharmaceutical development by emphasizing process understanding rather than relying on end-product testing. In the synthesis of Active Pharmaceutical Ingredients (APIs), variability in reaction conditions such as temperature, solvent composition, and catalyst loading can significantly influence impurity formation and overall product quality. Therefore, implementation of QbD principles is essential for ensuring process robustness and consistent product performance, particularly during scale-up and technology transfer. This review examines the application of QbD in pharmaceutical process development with a focus on risk assessment, Design of Experiments (DoE), and design space establishment. Risk assessment tools enable identification of critical process parameters, while DoE facilitates systematic evaluation of interactions between variables and optimization of operating conditions. The role of Process Analytical Technology (PAT) in real-time monitoring and control of manufacturing processes is also discussed. Despite the advantages of QbD implementation, challenges such as experimental complexity, data management, and integration of advanced analytical tools remain significant. Nevertheless, continued advancements in data analytics, automation, and real-time monitoring technologies are expected to further strengthen QbD-based pharmaceutical manufacturing.
Pharmaceutical process development has traditionally relied on empirical approaches, commonly referred to as trial-and-error methods, where process parameters are optimized through repeated experimentation followed by end-product testing. Although this approach has been widely used, it often provides limited understanding of the relationship between process variables and product quality. Consequently, manufacturing processes developed using empirical methods may exhibit variability, reduced robustness, and challenges during scale-up and technology transfer [1-4].
These limitations are particularly critical in the synthesis of Active Pharmaceutical Ingredients (APIs), where chemical reactions are highly sensitive to variations in process parameters such as reaction temperature, solvent composition, catalyst loading, mixing efficiency, and reaction time. Minor changes in these variables may significantly influence reaction kinetics, impurity formation, polymorphic behavior, and overall product yield. As a result, maintaining consistent product quality during scale-up becomes challenging without comprehensive process understanding [5-8].
To address these limitations, the pharmaceutical industry has adopted the Quality by Design (QbD) approach, which emphasizes systematic process understanding and risk-based development strategies. QbD focuses on building quality into the product during development rather than relying solely on end-product testing. This paradigm shift has been strongly encouraged by regulatory authorities through guidelines such as ICH Q8 (Pharmaceutical Development), ICH Q9 (Quality Risk Management), ICH Q10 (Pharmaceutical Quality System), and ICH Q11 (Development and Manufacture of Drug Substances) [1-4]. These guidelines promote a science-based framework that integrates risk assessment, statistical experimentation, and lifecycle management to ensure consistent product quality.
In the QbD framework, development begins with defining the Quality Target Product Profile (QTPP), followed by identification of Critical Quality Attributes (CQAs) that influence product safety and efficacy. Subsequently, Critical Material Attributes (CMAs) and Critical Process Parameters (CPPs) are identified through risk assessment and experimental studies. Statistical tools such as Design of Experiments (DoE) are then used to evaluate interactions between variables and optimize process conditions. The resulting knowledge enables the establishment of a design space within which product quality can be consistently maintained [9-14].
Although QbD provides significant advantages, implementation may present challenges including increased experimental complexity, requirement for statistical expertise, and large data management demands. Despite these challenges, QbD has become an essential framework for improving pharmaceutical manufacturing efficiency, ensuring regulatory compliance, and enhancing product quality [5–8].
This review aims to provide a comprehensive overview of Quality by Design implementation in pharmaceutical process development, with particular emphasis on risk assessment methodologies, Design of Experiments (DoE), design space establishment, and control strategy development for API synthesis.
QUALITY BY DESIGN FRAMEWORK IN API DEVELOPMENT

Quality by Design (QbD) represents a systematic, science-based, and risk-oriented approach to pharmaceutical development in which product quality is built into the manufacturing process through a thorough understanding of material attributes and process parameters.
Unlike traditional approaches that rely primarily on end-product testing, QbD emphasizes proactive identification and control of variables that influence product quality throughout the development lifecycle [1-4].
In Active Pharmaceutical Ingredient (API) synthesis, variability in raw materials, reaction conditions, and equipment performance can significantly affect Critical Quality Attributes (CQAs) such as purity, impurity profile, particle size, and polymorphic form. The QbD framework addresses these challenges by systematically identifying sources of variability and establishing control strategies that ensure consistent product quality. This structured approach improves process understanding, enhances manufacturing robustness, and facilitates regulatory flexibility during process optimization and scale-up [5-8].
The implementation of QbD in pharmaceutical development typically follows a structured sequence that includes defining the Quality Target Product Profile (QTPP), identifying CQAs, evaluating Critical Material Attributes (CMAs) and Critical Process Parameters (CPPs), performing risk assessment, applying Design of Experiments (DoE), establishing design space, and developing a comprehensive control strategy. This systematic methodology enables manufacturers to develop robust processes capable of consistently producing high-quality APIs [9-12].

1. Quality Target Product Profile (QTPP)
The Quality Target Product Profile (QTPP) is a prospective summary of the quality characteristics that a drug product should possess to ensure safety and efficacy. QTPP serves as a foundation for pharmaceutical development by defining desired attributes such as identity, strength, purity, stability, and dosage form [1,5]. In the context of API synthesis, QTPP may include parameters such as impurity limits, polymorphic form, particle size distribution, and chemical purity.
Establishing QTPP at the early stage of development allows systematic identification of attributes that must be controlled during manufacturing. This approach supports the development of a robust manufacturing process and facilitates regulatory acceptance of process design decisions [6,7].
2. Critical Quality Attributes (CQAs)
Variations in CQAs may affect safety, efficacy, and stability of pharmaceutical products. Therefore, identifying CQAs and understanding their relationship with process variables is essential for developing a reliable manufacturing process. This understanding enables manufacturers to control process variability and maintain consistent product quality [6-8].
Critical Material Attributes (CMAs) refer to the physical or chemical properties of raw materials, intermediates, or reagents that may influence product quality. Examples include purity of starting materials, particle size, moisture content, and solvent composition [2,6].
Critical Process Parameters (CPPs) are process variables that significantly impact CQAs and must be controlled within defined limits. In API synthesis, CPPs may include reaction temperature, reaction time, pH, mixing speed, and catalyst concentration. Identification of CMAs and CPPs is typically performed through risk assessment and experimental studies, which help determine how variations in process conditions influence product quality [9-12].
Design space refers to the multidimensional combination of input variables and process parameters that have been demonstrated to ensure consistent product quality. Establishing design space allows manufacturers to operate within defined limits without compromising product quality. This concept provides flexibility during manufacturing and supports regulatory acceptance of process adjustments [1,9].
Design space is typically established using statistical tools such as Design of Experiments (DoE) and response surface methodology. These approaches help identify optimal operating conditions and determine acceptable ranges for process parameters [10-12].
Lifecycle management is an essential component of QbD and involves continuous monitoring and improvement of the manufacturing process throughout the product lifecycle. Data generated during process development, scale-up, and commercial manufacturing are analyzed to evaluate process performance and identify opportunities for improvement [3,5].
Lifecycle management supports continued process verification and ensures that manufacturing processes remain robust over time. This approach enhances product quality, reduces variability, and supports regulatory compliance.
RISK ASSESSMENT IN API PROCESS DEVELOPMENT
Risk assessment is a fundamental component of the Quality by Design (QbD) framework and plays a critical role in identifying process variables that may influence product quality. In pharmaceutical development, risk assessment provides a structured approach to identifying, analyzing, and controlling potential sources of variability that may affect Critical Quality Attributes (CQAs). This systematic methodology is recommended in ICH Q9 guidelines, which emphasize risk-based decision-making throughout pharmaceutical development and manufacturing [2,3].
In Active Pharmaceutical Ingredient (API) synthesis, multiple variables such as raw material quality, reaction conditions, equipment performance, and environmental factors may influence product quality. Risk assessment enables identification and prioritization of these variables based on their potential impact. This approach allows researchers to focus on critical parameters during process development, improving efficiency and process robustness [5-8].
1. Risk Identification
Risk identification is the first step in the risk assessment process and involves determining potential sources of variability within the manufacturing process. In API synthesis, risk factors may arise from raw materials, process parameters, equipment performance, environmental conditions, and analytical measurement methods [6–8].
Common risk identification tools include:
The Ishikawa diagram is widely used in pharmaceutical development to categorize potential risk factors into major groups such as materials, methods, equipment, environment, personnel, and measurement. This structured approach helps identify sources of variability that may affect CQAs [15].
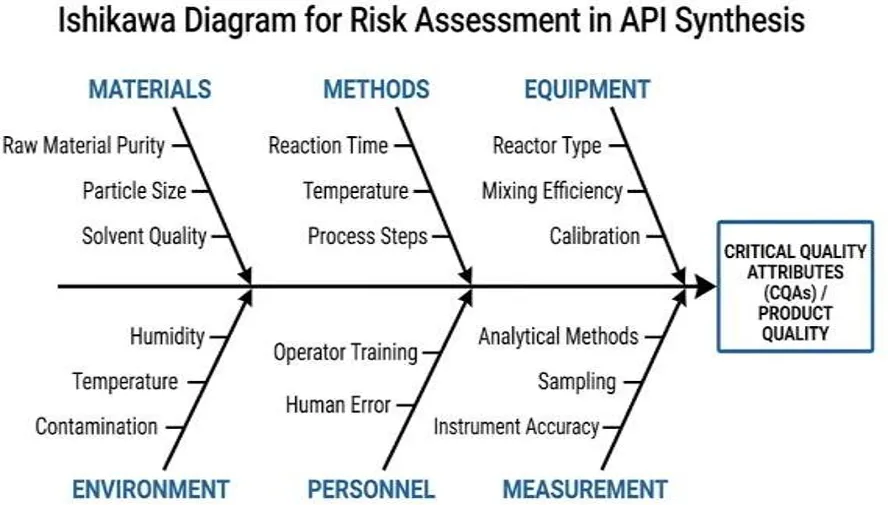
2. Risk Analysis and Evaluation
After identifying potential risks, the next step is risk analysis and evaluation. One commonly used method in pharmaceutical development is Failure Mode and Effects Analysis (FMEA).
This method evaluates risks based on three parameters:
These parameters are multiplied to calculate the Risk Priority Number (RPN), which is used to rank risks according to their potential impact [16].
The Risk Priority Number is calculated as:
RPN = Severity × Occurrence × Detectability
Variables with higher RPN values are considered critical and require further investigation using experimental approaches such as Design of Experiments (DoE) [12-14].
Table 1: Example FMEA Risk Assessment for API Synthesis
|
Process Step |
Risk Factor |
Impact |
Severity |
Occurrence |
Detectability |
RPN |
|
Reaction |
Temperature variation |
Impurity formation |
8 |
5 |
4 |
160 |
|
Raw material |
Impurity in input |
Reduced purity |
9 |
4 |
5 |
180 |
|
Mixing |
Poor agitation |
Incomplete reaction |
7 |
3 |
5 |
105 |
|
Solvent |
Incorrect ratio |
Yield reduction |
6 |
4 |
4 |
96 |
3. Risk Control and Mitigation
Risk control involves implementing strategies to reduce or eliminate identified risks. This may include adjusting process parameters, improving raw material specifications, or implementing in-process monitoring systems. In many cases, Design of Experiments (DoE) studies are conducted to understand the relationship between process variables and CQAs [12-14].
Effective risk control improves process robustness, reduces variability, and ensures consistent product quality. This approach also supports regulatory expectations and facilitates smoother process scale-up and commercialization [1-4].
DESIGN OF EXPERIMENTS (DOE) IN API PROCESS DEVELOPMENT
Design of Experiments (DoE) is a statistical methodology widely used in the Quality by Design (QbD) framework to evaluate the relationship between process variables and product quality attributes. Unlike traditional experimental approaches that vary one factor at a time, DoE allows simultaneous evaluation of multiple variables and their interactions. This approach improves process understanding and helps identify critical process parameters that influence Critical Quality Attributes (CQAs) [9-12].
In Active Pharmaceutical Ingredient (API) synthesis, variables such as reaction temperature, solvent composition, catalyst loading, reaction time, and mixing conditions may significantly affect yield and impurity formation. DoE provides a structured approach to evaluate these variables systematically, enabling optimization of process conditions and development of robust manufacturing processes [5-8].
1. Screening Experimental Designs
Screening experimental designs are used in early stages of process development to identify significant variables among multiple process parameters. These designs require fewer experiments and help narrow down the variables that significantly impact CQAs [9–12].
Common screening designs include:
These designs allow efficient identification of critical variables and reduce experimental workload during early-stage development [10-12].
Once significant variables are identified, optimization designs are used to determine optimal operating conditions. These designs evaluate both individual and interaction effects of process parameters [9–12].
Common optimization designs include:
These designs are often applied using Response Surface Methodology (RSM), which helps identify optimal process conditions and establish design space [10-12].
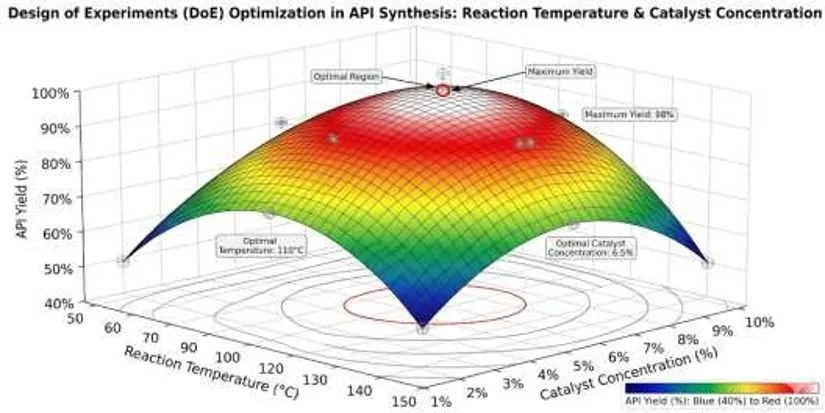
3. Statistical Modeling and Data Analysis
Data generated from DoE studies are analyzed using regression models. A quadratic regression equation commonly used in response surface modeling is:
Y = β₀ + β₁X₁ + β₂X₂ + β₁₂X₁X₂ + β₁₁X₁² + β₂₂X₂²
where:
Y = response variable (yield, impurity level)
X₁ and X₂ = independent variables
β = regression coefficients
Analysis of variance (ANOVA) is used to evaluate statistical significance of variables and validate model adequacy [10-12].
Table 2: Example DoE Matrix for API Optimization
|
Run |
Temperature (°C) |
Catalyst (%) |
Time (hr) |
Yield (%) |
|
1 |
50 |
1.0 |
3 |
82 |
|
2 |
60 |
1.0 |
3 |
86 |
|
3 |
50 |
2.0 |
3 |
88 |
|
4 |
60 |
2.0 |
3 |
92 |
|
5 |
55 |
1.5 |
4 |
90 |
4. Role of DoE in Design Space Development
One of the major outcomes of DoE studies is the establishment of design space. By analyzing response surfaces, researchers can determine acceptable ranges for process parameters.
Operating within the design space ensures consistent product quality and provides manufacturing flexibility [1,9-12].
DESIGN SPACE DEVELOPMENT AND PROCESS OPTIMIZATION
Design space is a key concept in the Quality by Design (QbD) framework and is defined as the multidimensional combination of input variables and process parameters that have been demonstrated to ensure consistent product quality. Establishing design space provides flexibility in manufacturing operations while maintaining product quality within predefined specifications. Regulatory agencies recognize that operating within an approved design space does not require additional regulatory approval, thereby improving manufacturing efficiency and process robustness [1-4].
In Active Pharmaceutical Ingredient (API) synthesis, design space development is achieved through systematic experimental studies and statistical modeling. Data obtained from Design of Experiments (DoE) studies are used to identify optimal operating conditions and acceptable ranges for critical process parameters. This approach improves process understanding and reduces variability during scale-up and commercial manufacturing [9-12].
The establishment of design space begins with identifying Critical Process Parameters (CPPs) and Critical Material Attributes (CMAs) that influence Critical Quality Attributes (CQAs).
These parameters are typically identified through risk assessment and screening experiments [2,5-8].
Common variables evaluated in API synthesis include:
Each parameter is evaluated to determine its effect on CQAs such as yield, purity, and impurity profile. Parameters with significant influence are selected for further optimization during design space development [9-12].
Design space is typically represented as a multidimensional region defined by combinations of process variables that produce acceptable product quality. Statistical tools such as response surface methodology are used to visualize this region using contour plots or three-dimensional response surfaces [10-12].
For example, in API synthesis, interaction between reaction temperature and catalyst concentration may significantly affect product yield and impurity formation. By analyzing response surface plots, acceptable operating ranges can be established to maintain product quality within defined limits [9-12].
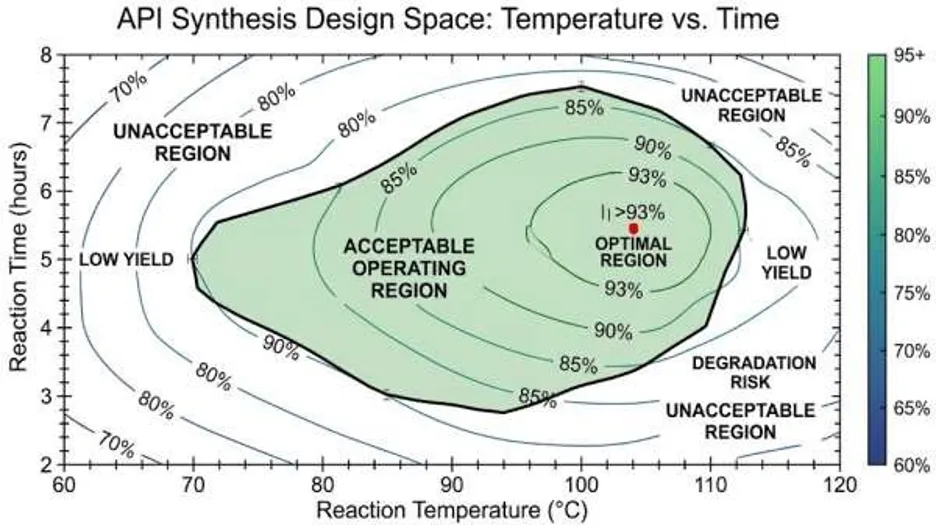
Figure 5: Design Space Contour Plot
The concept of design space provides regulatory flexibility in pharmaceutical manufacturing. Regulatory authorities recognize that adjustments within the approved design space do not require additional regulatory submissions, provided product quality remains unaffected [1-4].
This flexibility allows manufacturers to optimize process conditions, accommodate variability in raw materials, and improve manufacturing efficiency while maintaining consistent product quality [5-8].
Design space development improves process understanding and supports robust manufacturing. By defining acceptable operating ranges, manufacturers can reduce variability, minimize batch failures, and enhance process reliability. Furthermore, design space facilitates technology transfer from laboratory-scale development to commercial manufacturing [9-12].
CONTROL STRATEGY AND PROCESS MONITORING IN API MANUFACTURING
Control strategy is a critical component of the Quality by Design (QbD) framework and refers to a planned set of controls implemented to ensure consistent product quality throughout the manufacturing process. A well-defined control strategy is developed based on knowledge gained from risk assessment, Design of Experiments (DoE), and design space studies. This approach ensures that Critical Quality Attributes (CQAs) remain within acceptable limits during manufacturing [1-4].
In Active Pharmaceutical Ingredient (API) synthesis, control strategies involve monitoring raw materials, process parameters, and product quality throughout the manufacturing lifecycle. These controls improve process reliability, reduce variability, and support regulatory compliance [5-8].
Raw material quality plays an important role in API synthesis. Variability in raw materials such as starting material purity, particle size, and moisture content may influence reaction performance and product quality. Therefore, raw material specifications must be carefully defined and controlled [5-8].
Common raw material controls include:
Implementing these controls reduces variability and improves process consistency.
In-process controls involve monitoring process parameters during manufacturing to ensure product quality. These controls enable early detection of deviations and allow corrective actions before product quality is affected [3,5].
Common in-process parameters monitored during API synthesis include:
Analytical techniques such as chromatography and spectroscopy are commonly used for in-process monitoring [17].
3. Process Analytical Technology (PAT)
Process Analytical Technology (PAT) enables real-time monitoring of manufacturing processes and supports process control within the design space. PAT tools provide continuous measurement of process variables and help detect deviations during production [5,17].
Common PAT tools include:
PAT implementation improves process understanding and supports real-time decision-making.
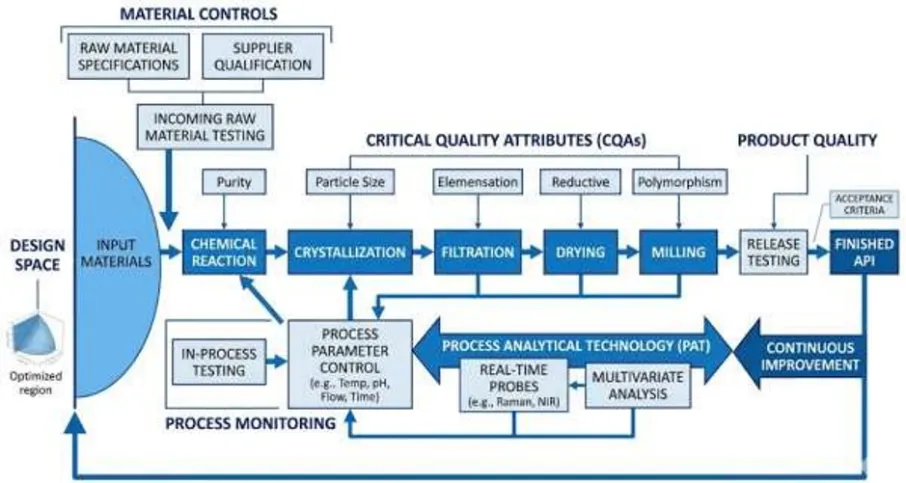
Figure 6: Control Strategy Framework in QbD
Real-time monitoring integrates analytical tools with automated process control systems. This approach allows continuous monitoring of process parameters and ensures operation within design space limits [5].
Real-time monitoring also supports Real-Time Release Testing (RTRT), where product quality is verified using process data instead of final product testing. This reduces production time and improves efficiency [5,17].
Lifecycle management involves continuous monitoring and improvement of manufacturing processes throughout the product lifecycle. Data collected during commercial manufacturing are analyzed to identify trends and improve process performance [3,5].
Lifecycle management ensures consistent product quality and supports regulatory expectations for continuous process verification.
CHALLENGES IN IMPLEMENTATION OF QUALITY BY DESIGN IN API MANUFACTURING
Although Quality by Design (QbD) provides a structured and science-based framework for pharmaceutical development, its implementation in Active Pharmaceutical Ingredient (API) manufacturing presents several technical, operational, and regulatory challenges. The transition from traditional empirical development to a QbD based approach requires significant investment in resources, multidisciplinary expertise, and advanced analytical tools. These challenges may affect the pace of adoption, particularly in small and medium-scale pharmaceutical industries [5-8].
One of the primary challenges associated with QbD implementation is the increased complexity of experimental design. Unlike traditional approaches that rely on one-factor-at-a-time experimentation, QbD requires systematic evaluation of multiple variables simultaneously. This often involves extensive Design of Experiments (DoE) studies, which may require a large number of experimental runs depending on the number of variables involved [9-12].
For complex API synthesis processes, multiple variables such as temperature, solvent composition, catalyst loading, mixing speed, and reaction time must be evaluated. As the number of variables increases, the experimental workload grows significantly, leading to increased time, material consumption, and cost. These challenges may delay early-stage process development and increase overall project timelines [6-8].
Successful implementation of QbD requires collaboration among professionals from multiple disciplines, including process chemistry, analytical chemistry, quality assurance, regulatory affairs, and statistical modeling. The use of advanced statistical tools such as response surface methodology and multivariate analysis requires specialized expertise in data interpretation and modeling [10-12].
Many pharmaceutical development teams may lack adequate statistical knowledge, making it difficult to design and interpret DoE studies effectively. As a result, organizations may need to invest in training programs or recruit experts with specialized knowledge in statistical process design and data analytics [6,8].
QbD-based process development generates large volumes of experimental and analytical data. Managing, analyzing, and interpreting this data can be challenging, especially when multiple process variables and interactions are involved. Proper data management systems and statistical software are essential for handling complex datasets and developing predictive models [9-12].
Without robust data analysis systems, there is a risk of misinterpreting experimental results, which may lead to incorrect identification of critical process parameters. Additionally, maintaining data integrity and traceability is essential to meet regulatory requirements and support decision-making during process development [3,5].
Process Analytical Technology (PAT) is an important component of QbD that enables real-time monitoring of manufacturing processes. However, implementation of PAT tools requires significant investment in analytical instruments, software systems, and process integration.
Instruments such as Raman spectroscopy, near-infrared spectroscopy, and online chromatography require validation, calibration, and maintenance [5,17].
In addition, integration of PAT tools with existing manufacturing systems may require modifications in process equipment and automation infrastructure. These factors may increase implementation costs and complexity, particularly for smaller pharmaceutical companies [5,8].
Implementation of QbD often requires a shift in organizational mindset. Traditional pharmaceutical development approaches rely heavily on fixed process conditions and end-product testing. In contrast, QbD promotes a knowledge-based and flexible development strategy focused on process understanding and continuous improvement [3,6].
This transition may require changes in internal processes, employee training, and quality management systems. Resistance to change and lack of awareness about QbD benefits may slow down adoption within organizations [6-8].
Although regulatory agencies encourage QbD implementation, they also require detailed documentation of development studies. Regulatory submissions must include comprehensive descriptions of risk assessment, DoE studies, design space justification, and control strategies. Preparing such documentation requires significant effort and detailed reporting of experimental results [1–4].
Furthermore, regulatory expectations may vary across regions, which may complicate global product development strategies. Ensuring compliance with multiple regulatory frameworks requires careful planning and documentation [3,5].
Implementation of QbD requires investment in experimental studies, advanced analytical tools, and trained personnel. These requirements may increase development costs, particularly during early stages of product development. Small and medium pharmaceutical companies may face resource constraints that limit full implementation of QbD principles [6-8].
Despite these challenges, long-term benefits of QbD implementation include improved process robustness, reduced batch failures, and enhanced product quality, which may offset initial investment costs.
Table 3: Challenges in QbD Implementation
|
Challenge |
Description |
Impact |
|
Experimental complexity |
Large number of experiments |
Increased development time |
|
Statistical expertise |
Need for advanced modeling |
Training requirements |
|
Data management |
Large datasets |
Complex analysis |
|
PAT implementation |
Instrument cost |
High investment |
|
Organizational change |
Cultural shift |
Slow adoption |
|
Regulatory documentation |
Detailed submission |
Increased workload |
FUTURE PERSPECTIVES IN QBD-BASED PHARMACEUTICAL MANUFACTURING
Quality by Design (QbD) has significantly transformed pharmaceutical development by promoting a systematic and science-based approach to process design and control. As pharmaceutical manufacturing continues to evolve, emerging technologies such as advanced data analytics, artificial intelligence, continuous manufacturing, and digital process monitoring are expected to further enhance the effectiveness of QbD implementation. These advancements aim to improve process understanding, reduce variability, and ensure consistent production of high-quality pharmaceutical products [5-8].

1. Integration of Advanced Data Analytics
The increasing availability of large datasets from process development and manufacturing operations has created opportunities for advanced data analytics in pharmaceutical development. Modern data analysis techniques such as multivariate data analysis (MVDA) and predictive modeling can improve process understanding and identify complex relationships between process parameters and product quality attributes [18-20].
Advanced data analytics also supports real-time process optimization and predictive process control. By analyzing historical process data, manufacturers can identify trends, detect deviations, and implement corrective actions proactively. These approaches contribute to improved process robustness and reduced manufacturing risks [18-20].
2. Artificial Intelligence and Machine Learning Applications
Artificial intelligence (AI) and machine learning (ML) are emerging as powerful tools in pharmaceutical development and manufacturing. These technologies enable automated analysis of large datasets and identification of hidden relationships between process variables and product quality [18-20].
Machine learning models can be used to predict reaction outcomes, optimize process parameters, and enhance process control strategies. AI-based predictive modeling can also support risk assessment, design space development, and real-time process monitoring.
Integration of AI with QbD frameworks is expected to significantly reduce development time and improve process efficiency [18-20].
3. Continuous Manufacturing
Continuous manufacturing is gaining increasing attention in pharmaceutical production due to its potential to improve process efficiency and product quality. Unlike traditional batch manufacturing, continuous manufacturing allows continuous feeding of raw materials and simultaneous production of finished products. This approach enables better process control and reduced variability [20].
Continuous manufacturing aligns with QbD principles by enabling real-time monitoring and control of process variables. Regulatory agencies have also encouraged the adoption of continuous manufacturing as part of modern pharmaceutical manufacturing strategies. The integration of continuous manufacturing with QbD is expected to improve manufacturing efficiency and product consistency [5,20].
4. Real-Time Release Testing
Real-time release testing (RTRT) is an important advancement associated with QbD implementation. RTRT involves evaluating product quality based on real-time process monitoring data instead of relying solely on final product testing. This approach is supported by Process Analytical Technology (PAT) tools and automated data analysis systems [5,17].
Real-time release testing reduces production delays and improves manufacturing efficiency. Additionally, RTRT supports continuous process verification and enhances product quality assurance [5,17].
5. Digitalization and Smart Manufacturing
Digitalization and smart manufacturing technologies are transforming pharmaceutical production systems. Advanced sensors, automation systems, and digital monitoring platforms enable continuous data collection and real-time process control. These technologies support predictive maintenance, automated decision-making, and improved process efficiency [18–20].
Smart manufacturing also facilitates integration of QbD principles with Industry 4.0 technologies. This integration is expected to enhance process understanding, reduce variability, and improve overall pharmaceutical manufacturing performance.
CONCLUSION
Quality by Design (QbD) has emerged as a systematic and science-based framework for improving pharmaceutical process development and manufacturing. By shifting the focus from end-product testing to process understanding, QbD enables identification and control of variables that influence product quality. In Active Pharmaceutical Ingredient (API) synthesis, implementation of QbD principles facilitates improved process robustness, reduced variability, and consistent production of high-quality pharmaceutical products.
The integration of risk assessment methodologies, Design of Experiments (DoE), and design space development allows systematic identification of Critical Quality Attributes (CQAs), Critical Material Attributes (CMAs), and Critical Process Parameters (CPPs). These approaches enhance process understanding and support optimization of manufacturing conditions. Furthermore, the development of comprehensive control strategies, supported by Process Analytical Technology (PAT) and real-time monitoring, ensures consistent product quality throughout the manufacturing lifecycle.
Despite the advantages associated with QbD implementation, several challenges remain, including experimental complexity, data management requirements, and the need for multidisciplinary expertise. However, ongoing advancements in artificial intelligence, data analytics, continuous manufacturing, and digital process monitoring are expected to further strengthen QbD-based pharmaceutical manufacturing.
Overall, the adoption of QbD principles provides a robust framework for improving pharmaceutical development, enhancing regulatory flexibility, and ensuring consistent production of high-quality APIs. Continued integration of advanced technologies and data-driven approaches will play a crucial role in advancing QbD-based pharmaceutical manufacturing in the future.
ACKNOWLEDGMENT
The authors would like to acknowledge the support and facilities provided by the Department of Pharmaceutical Quality Assurance, Rashtrasant Janardhan Swami College of Pharmacy, Kokamthan, Taluka Kopargaon, District Ahmednagar, Maharashtra, India, for the successful completion of this work. The authors express their sincere gratitude to Mr. Dadasaheb Kawade for his valuable guidance and continuous support throughout the preparation of this manuscript. The authors also extend their sincere thanks to Dr. Vijay Jadhav, Head of the Department, for his encouragement and support. The authors further acknowledge Dr. Nitin Jain, Principal, and Dr. Nitin Jain, vice-principal for providing the necessary facilities and institutional support to carry out this work.
ABBREVIATIONS
REFERENCES

Tanmayi Jadhav, Dadasaheb Kawade, Dr. Vijay Jadhav, Dr. Nitin Jain, Dr. Usha Jain, Implementation of Quality by Design (QbD) in Pharmaceutical Process Development: Risk Assessment, Design of Experiments, and Control Strategy for API Synthesis, Int. J. of Pharm. Sci., 2026, Vol 4, Issue 7, 826-842. https://doi.org/10.5281/zenodo.21176761
 10.5281/zenodo.21176761
10.5281/zenodo.21176761